Review of Volatile Perfluorocarboxylic Acids as Ion Pair Reagents in LC: Part I
LCGC North America
This article facilitates the selection of various perfluorocarboxylic acids as ion-pair reagents in LC.
Dolan (1) outlined in LCGC the complicating factors of ion pairing in reversed-phase liquid chromatography (LC), and reported that ion pairing is less popular today than in the past. The concentration of reagent on the stationary phase is controlled by several variables, including the organic content of the mobile phase and the column temperature. Slow equilibration between mobile phase and column complicates gradient elution, and ion-pair reagents are best used with a dedicated column.
However, volatile perfluorocarboxylic acids of longer n-alkyl chain (instead of trifluoroacetic acid [TFA]) are increasingly popular as ion-pair reagents because of their compatibility with LC and UV (2–7), fluorescence (8–12), Fourier-transform infrared (FT-IR) (13), radiochemical (14), electrochemical (15–19), electrospray ionization mass spectrometry (ESI-MS) (most of the reviewed publications), inductively coupled plasma (ICP)–MS (20–22), matrix-assisted laser desorption ionization (MALDI) (23,24), or evaporative light scattering (ELS) (18), (25–31) detection. However, these perfluorinated surfactants can cause serious environmental problems if not disposed of properly (32–36). Figure 1 shows the number of publications since 1998 that use these reagents in LC. The recent increase documented in this review is perhaps due to reduced ionization suppression in ESI-MS as well as improved resolution, compared with TFA.


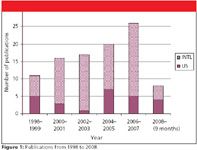
Figure 1
Perfluorocarboxylic acids have been used in the analysis of cephalosporin (37), aminoglycosides (9,15,16,18,19,29,30,38–45), amoxicillin (46), spectinomycin (47), catecholamines (48–50), acetylcholine (51), quaternary ammonium herbicides (52–55) and pesticides (56), underivatized amino acids (5,17,25,28,57–69), peptides (5,8,23,24,63,70–75), proteins (2,13,76–80), monoclonal antibodies (81), epibatidine (82), 1-aminocyclopropane-1-carboxylic acid (83), amines (77,84), carnitine and acylcarnitine (85–88), siderophores (89), selenomethionine (20–22,90,91), betacyanins (3), saxitoxin (92,93), vitamins (94), metals in food supplements and pharmaceuticals (26), methadone (95), serine protease inhibitors (96), triethylenetetramine (97), flavonoids and alkaloids (6), glutamine and GABA (98), mosapride (99), amodiaquine (100), pyridinoline (10,11), tetrodotoxin (12), desmosine (101), diallyldimethylammonium chloride (102), and rat serum esterases (14). This review discusses the following perfluorocarboxylic acids: n-pentafluoropropionic acid (PFPA), n-heptafluorobutyric acid (HFBA), nonafluoropentanoic acid (NFPA), tridecafluoroheptanoic acid (TDFHA), and pentadecafluorooctanoic acid (PDFOA).
Reagent Quality
The purity and volatility of these reagents are important. For example, PDFOA is manufactured by either an electrochemical fluorination (ECF) or a telomerization process (34); both processes have perfluorocarboxylate impurities present. A typical batch of PDFOA made by the ECF process can contain 22% by weight branched isomers (103). In most cases, the vendor provides a percentage purity range, determined by titration, which might be inadequate for determining the effects of isomers on ion-pairing behavior in LC, and could compound problems with "system peaks" discussed later in this review. For example, in the LC analysis of PDFOA itself, a slow gradient can separate branched isomers completely from the linear isomer (34). Petritis and colleagues (104) noted that the purity of the perfluorocarboxylic acids ranged from 96% to 99% and varied by lot, even from the same supplier, but did not contain nitrogen contaminants. A commercial lot of PDFOA supplied to the authors tested as 95.4% purity by titration and 4.2% water, according to the vendor's specification sheet.
Petersson and colleagues (105) attempted to investigate the C6–C12 homologs of perfluorocarboxylic acids in micellar electrokinetic chromatography, but had problems with reagent purity. Three different batches of perfluorononanoic acid from two suppliers contained brown particles and gave colored solutions as well as large differences in selectivity and retention. Undecafluorohexanoic acid and TDFHA gave large differences in current and retention between solutions prepared from the same batch.
James and colleagues (10) used HFBA in LC of pyridinoline with native fluorescence detection, but trace fluorophores in HFBA, present to varying levels depending upon the synthetic batch, made its further purification essential. Tsuji and colleagues (106) found it essential to purify perfluoro-n-alkanoic acids (C8–C18) by repeated recrystalizations from n-hexane–acetone mixed solvent, before measurement of their physico-chemical properties. Using gas chromatography (GC)–MS, 19F nuclear magnetic resonance (NMR), and elemental analysis, they found purity after recrystallization to be greater than 99.5%, but made no mention of branched isomers in these samples.
Generally, electrolyte volatility is estimated only through the boiling point of the mobile phase additives, the mass spectrometry "pollution" and long-term signal stability of test solutes. However, Elfakir's group (27) tested eluent volatility with ELSD, because this detection technique requires a volatile mobile phase to avoid high background noise. PDFOA had a limit of solubility of 5 mM in water, and all the perfluorocarboxylic acids tested were at least volatile at 2 mM. However, almost all combinations of these acids with bases (even ammonia) resulted in "nonvolatile salts." However, low concentrations (for example, 0.5 mM of PDFOA) were enough to provide retention and high LC selectivities among very polar underivatized amino acids. Therefore, ammonium salts of PDFOA and shorter alkyl chain homologues at concentrations well below 2 mM appear to be volatile enough for ESI-MS ("buffered" mobile phases are reported frequently) where the evaporation conditions employ a much higher temperature and vacuum.
Which of these acids should be chosen and at what concentration and pH? Should samples be prepared in mobile phase, or will they be unstable under such acidic conditions? If elevated temperature and gradient elution are used, will there be problems with lengthy equilibration times, "system peaks," irreproducible retention times, column stability up to 90 °C with acidic mobile phases and prolonged use, and frequent column flushing? If ESI-MS detection is used, how much ionization suppression will occur, depending upon the acid chosen, its concentration, and the design of the ESI source? Will this method be robust and transferable between laboratories?
Selectivity and Choice of Ion-Pair Reagent
Almost 30 investigators (Table I) compared several of the acids for their applications before deciding on the best choice, but most did not report a reason for their selection, other than occasionally referencing a similar application. There are, however, patterns in the choice of these reagents, based upon the analytes, chronology, and detection method. Investigators who compared these reagents chose HFBA, NFPA, and PDFOA; 61%, 32%, and 7% of the time, respectively. HFBA was favored for proteins and peptides, for example, while PDFOA had selectivity advantages for amino acids. Generally, the longer the alkyl chain of the acid and the higher the concentration, the longer the retention time was, usually with improved peak shape. However, tradeoffs against column equilibration and run time were made. Frequent changes in elution order of several analytes occurred when longer alkyl chain acids were substituted for shorter alkyl chain homologs (21,22,25,28,30,63,68), or when reagent concentrations (4,7) or column temperatures (48,52) were increased, as described in the following individual reagent sections.


Table I: Publications comparing selectivity of perfluorocarboxylic acids
PDFOA
PDFOA has not been selected since 2002 (Table II), but has been compared with its shorter alkyl chain homologs as recently as 2008 (96). From 1998 to 2002, PDFOA was selected a total of five times; four times for amino acids (25,28,57,59), and once for proteins (73). For very polar compounds, PDFOA can offer retention and selectivity advantages and lower ionization suppression in MS analyses. However, it has disadvantages of longer equilibration times, retention time drift, and "system peaks." Shen and colleagues (96) reported PDFOA formed esters with methanol in the mobile phase at room temperature over several days. Although PDFOA (solid) gave the best sensitivity of five perfluorocarboxylic acids tested, NFPA (liquid) was chosen, because it gave adequate sensitivity, was convenient to dispense, and reacted more slowly with methanol. However, Snoble and colleagues (109) documented that even dilute formic acid solutions in methanol decline in acid content and form methyl formate within two days at room temperature. This mobile phase degradation was sufficient to shift retention times of test peptides slightly and change their resolution.

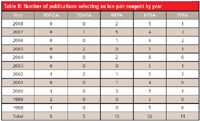
Table II: Number of publications selecting an ion-pair reagent by year
Petritis and colleagues (25,28) obtained better sensitivities for amino acids at higher PDFOA concentrations, but the analysis time was increased significantly. Selectivity was sometimes better at low concentration, but the elution order was always the same, whatever the concentration of PDFOA. However, with some reagents such as TDFHA, the elution order of some amino acid pairs inverted with increasing TDFHA concentration. Using 10 polar amino acids, they studied retention factor versus reagent concentration. HFBA and NFPA were not efficient enough to separate the 10 amino acids, but PDFOA had higher selectivities, two to three times those of HFBA and NFPA.
Chaimbault and colleagues (57) found that under isocratic conditions, basic amino acids were retained highly with 0.5 mM PDFOA; therefore, a gradient elution was used, based upon simultaneously increasing the concentration of acetonitrile and decreasing the reagent concentration. Ishihama and colleagues (73) claimed that PDFOA had hydrophobicity and ion-pairing capabilities similar to sodium dodecyl sulfate (SDS). In all these publications, the PDFOA concentration was usually 0.5 mM, and once it was 1 mM, with one notable exception (73), which used 12.5 mM, most likely 10 times what was needed. In general, at low ion-pair concentration there is roughly a linear increase in retention with reagent concentration. At high reagent concentration, the increase in retention levels off due to saturation of the stationary phase with reagent. This relationship depends upon the chain length (hydrophobicity) of the reagent, mobile phase composition, temperature, and pH.
TDFHA
TDFHA was selected a total of five times since 2001, most recently as 2007; four times for amino acids (60,62,67,69), and once for proteins and peptides (71). Gustavsson and colleagues (77) found TDFHA gave worse repeatability and broader peaks than HFBA for several amines. Petritis and colleagies (71) evaluated 0.5–5 mM TDFHA, HFBA, NFPA, and PDFOA for underivatized small peptides. HFBA did not separate the most polar peptides and PDFOA gave the highest retention, but not the best selectivity. NFPA and TDFHA were the most suitable, but gradient elution was required to elute the less polar peptides. In all of these publications, concentrations of TDFHA ranged 0.5–3 mM, but mostly 0.5–1 mM.
NFPA
The 15 publications on NFPA are spaced evenly chronologically (except 2007, with five publications) and are more diverse than PDFOA and TDFHA, including five on amino acids (58,61,66,68,110), three on peptides and proteins (74,75,80), and two on aminoglycosides (29,30).
Chaimbault and colleagues (58) separated underivatized amino acids using a column packed with porous graphitic carbon and compared several of these reagents. PDFOA and TDFHA did not provide the best selectivity, even with increased concentration, and HFBA didn't give as good efficiency as 20 mM NFPA, but decreasing the column temperature to 10 °C improved the separation.
Kwon and colleagues (68) tested TFA, HFBA, and NFPA on a C18 microbore column with amino acids, but the separation efficiency of HFBA and TFA was not as good as NFPA. Gammelgaard and colleagues (20) used a C18 column to test 10 mM HFBA, 2 and 3 mM NFPA, and 0.2 mM TDFHA for the analysis of selenoamino acids in urine. In 10 mM HFBA, the analytes were coeluted, while 3 mM NFPA gave the best separation, and 0.2 mM TDFHA achieved separation, but the elution order was different. This difference in selectivity could be useful, but the solubility of TDFHA is low and problems with precipitation in urine samples were observed. The concentration of reagent necessary for species separation decreased from 10 mM HFBA to 0.2 mM TDFHA, and 3 mM NFPA was chosen.
Teerlink and colleagues (61) measured carboxymethyl lysine in human plasma. Analytes and standards were eluted as symmetric peaks, clearly resolved from interfering compounds with a 5 mM NFPA gradient. Galanakis and colleagues (30) compared formic acid and TFA, HFBA, and NFPA in the analysis of amikacin, using ELSD. Increased reagent concentration decreased the amikacin retention time, with the exception of NFPA. 18.2 mM NFPA was the most efficient reagent for the separation of amikacin from the closely related kanamycin.
In previous studies of histidine and peptides, Orioli and colleagues (75) used 7.7 mM HFBA, but later substituted 5 mM NFPA, because the internal standards were coeluted near the void volume, and matrix interferences were coeluted with other analytes. NFPA increased retention time and shifted the more lipophillic analytes beyond the matrix components. Spacil and colleagues (26) compared TFA, PFPA, and NFPA for the separation of sodium, magnesium, ascorbate, and citrate in pharmaceutical formulations. The retention of a metal cation was longer with NFPA than TFA, but the retention of ascorbate and citrate was unaffected. 3.2 mM NFPA was used for the first part of the chromatographic elution and sodium and potassium were separated from magnesium and calcium; however, separation of metal cations with equal charge was not possible.
Liu and colleagues (66) tested both 5 mm NFPA and TDFHA. TDFHA strongly retained nonpolar amino acids and, thus, required a higher concentration of acetonitrile to elute them. Polar and acidic amino acids were retained effectively by NFPA, but they were coeluted. LC gradient conditions were optimized to separate several critical pairs of amino acids, including isomeric leucine–isoleucine and isobaric glutamine–lysine. In all of these publications, the concentration of NFPA ranged 2–10 mM, with a few outliers at 20 mM, but usually around 5 mM.
Shen and colleagues (96) separated stereoisomers of a serine protease inhibitor, noting stereoselective resolution of the individual diastereomers increased, presumably by inducing conformational differences in the ion-pair complexes of each isomer that were sufficient to affect separation on a hydrophobic column. They evaluated TFA, HFBA, NFPA, TDFHA, and PDFOA using a sub-2-µm C18 column, and investigated 6.5–104 mM NFPA in methanol: water and elevated column temperature settings of 40–100 °C. Contrary to other publications, at a constant concentration (20 mM) of reagent, as the molecular weight of the reagent increased, the corresponding retention times decreased. As the concentration of NFPA increased from 6.5 to 104 mM, the resolution between the diastereomers increased (with little change in retention time), with no additional benefits for NFPA concentrations above 78 mM.
HFBA
HFBA is the most popular reagent, with nearly 60 publications and the most diverse applications. Peptide and protein analyses are the most frequent (thirteen), followed by aminoglycosides (seven), carnitine (four), selenium compounds (four), and herbicides (four). HFBA seems to be a compromise between shorter equilibration times (at temperatures around 30 °C) and selectivity compared with TDFHA and PDFOA.
Neubecker and colleagues (48) obtained adequate LC retention and excellent peak shape for norepinephrine with 10 mM HFBA, with no loss of instrument sensitivity over time. Castro and colleagues (52) improved peak shape of quaternary ammonium herbicides with increased PFPA and HFBA concentration. Because similar LC results were obtained with 15 mM HFBA and PFPA but HFBA gave the highest MS response, it was chosen over PFPA. They studied the effect of column temperature (C8 column) and found that at 50 °C, the elution order completely changed.
Kotrebai and colleagues (21 22), studying selenium compounds by ICP-MS, chose 0.1% TFA when a later eluted compound was of interest because, compared with 7.7 mM HFBA, it gave increased peak height and a better signal-to-noise ratio (S/N), but HFBA was chosen for early eluted compounds. HFBA was preferred over PFPA because of higher available purity and substantial retention time drift with PFPA. There were several compounds that switched elution order between TFA and HFBA. Lindemann and colleagues (91) compared TFA, PFPA, and HFBA and found that 7.5 mM HFBA (pH 3.5) exhibited optimal separation and retention of selenium compounds. Zhu and colleagues (51) tested HFBA, NFPA, and PDFOA, and all gave good separation of acetylcholine and choline; however, HFBA was chosen because it had a shorter equilibration time. Heller and colleagues (107) preferred HFBA in aminoglycoside analysis because it used more methanol compared with PFPA and TFA and, thus, improved the electrospray process. Hashimoto and colleagues (93) used a C30 column and 5 mM HFBA to separate saxitoxins in clam extracts from contaminants (matrix).
McCormack and colleagues (89) tested four different columns (C18, porous graphitic carbon, C8-amino, and polystyrene–divinylbenzene [PS–DVB]) for the analysis of siderophores, and found that 7.7 mM HFBA improved the response and retention of the iron(III) complex of rhodotoluric acid. Delgado and colleagues (2), using a PSDVB column at 50 °C, analyzed soya bean proteins, comparing TFA, morpholine, and HFBA and obtained the best results with mixtures of TFA and morpholine. Vernez and colleagues (85) noted above 10 mM HFBA, retention times of long chain acylcarnitines increased without any further benefit for the retention of carnitine and acylcarnitine. Dillon and colleagues (81) reported the combination of HFBA and TFA (0.4 mM and 0.05%, respectively) was critical for adequate resolution of monoclonal antibodies. TFA concentration was lowered without losing separation and provided unique resolution not possible with TFA or HFBA alone, while PFPA did not improve the separation.
Walcher and colleagues (70) used HFBA instead of TFA for protein analysis and increased retention and improved selectivity. Liu and colleagues (24) reported in the analysis of glycosylated peptides, the higher the number of amines in the peptide, the more ion pairing occurred, and longer retention times were observed. Surprisingly, HFBA separated the monophosphorylated peptides from the diphosphorylated analogs better than any other ion-pair reagent, including TFA. Wybraniec and colleagues (3), in an analysis of betacyanins, noted that HFBA and PFPA concentration had more effect upon retention than TFA, and markedly increased in the 0–5 mM range, but retention times shifted. Megoulas and colleagues (29) observed that retention of kanamycins A and B was highly dependent upon mobile phase acidity (TFA, trichloroacetic acid [TCA], and HFBA were compared) and increased acidity decreased retention times and resolution of both kanamycins. Midttun and colleagues (94) used 2.5 mM HFBA on a C8 column to retain the highly polar B6 species containing phosphate groups, whereas elution of the larger B2 species required an acetonitrile gradient.
Tholey and colleagues (63), using a capillary PS–DVB column and a gradient with 3.8 mM HFBA, found no correlation between the length of the peptide and overall retention. Hydrophobicity, and not analyte charge, was the primary criterion determining retention. TFA did not work well, but HFBA separated all six peptides. The elution order of the phospho- and the nonphosphopeptides was reversed compared to TFA. Shibue and colleagues (5) used 10, 20, and 30 mM HFBA on a C8 column and found the effect of increasing counter ion hydrophobicity was more marked the greater the positive charge on the peptides, whereas the effect of increasing the concentration was not so clear cut. Wang and colleagues (78) found 1.5 mM HFBA (versus TFA) significantly decreased the resolution of their model proteins.
Mant and colleagues (7) prepared two series of four synthetic peptide analogs ("a" and "b"), with both series having net charges of +1, +2, +3, and +4; the only difference between the two series was the substitution of one hydrophobic residue ("b" series) for a glycine residue in the peptide sequence. Both PFPA and HFBA were investigated and peptide retention time increased markedly both with the increasing peptide net charge and hydrophobicity of the "b" series. Elution order reversals occurred with varying reagent concentration.
Sun and colleagues (86) improved peak broadening and tailing problems with the basic amino groups of acylcarnitines with 3.8 mM HFBA. Initially, Naldi and colleagues (79) did not obtain resolution because TFA and HFBA were used at a concentration that was too high, increasing histone protein retention times and losing selectivity. A medium lipophilic column (C4) and lower concentrations of HFBA (3 mM) gave better selectivity than 0.4% formic acid. Xi and colleagues (15) tested three stationary phases for the analysis of etimicin impurities and found 7.4–15 mM HFBA had no significant effect on retention.
Gao and colleagues (95) compared HFBA, NFPA, TDFHA, and PDFOA added to the sample, and not to the mobile phase. At 6.3 mM NFPA in the sample, the retention time of methadone was similar to that of 6.3 mM TDFHA, but with significant differences between NFPA and PDFOA. No concentration of NFPA gave a similar retention, even with a saturated uptake of the reagent on the column. The addition of HFBA to the samples significantly reduced carryover.
Ding and colleagues (6) enhanced the retention of synephrine significantly with 9.6 mM HFBA, while also decreasing band tailing. Hakkinen and colleagues (84) studied underivatized polyamines and observed severe peak tailing with weaker acids, compared to stronger acids. Also, with water as sample dilution solvent, some of the compounds were not retained completely by the column. However, when 7.7 mM HFBA (that is, mobile phase) was used for dilution, all the compounds were retained. Lu and colleagues (97) studied HFBA, NFPA, and PFPA for the analysis of triethylenetetramine (TETA) in human plasma. 7.7 mM HFBA produced the best result, and also was used in the samples to stabilize analytes and reduce carryover.
Eckstein and colleagues (98) achieved better durability using 38.4 mM HFBA and 1% formic acid, versus PDFOA and TFA, for the separation of glutamine, glutamate, pyroglutamate, and GABA in cerebrospinal fluid (CSF). De Person and colleagues (64) selected 7 mM HFBA in a gradient elution analysis of native amino acids, but found TDFHA and PDFOA were better (compared with TFA and HFBA) for the separation on C8 and C18 columns.
Shibue and colleagues (4) compared 1–60 mM HFBA and PFPA on a C8 column. Peptides of differing charges moved at differing rates relative to each other, depending upon the concentration of reagents. Concentrations above 10 mM HFBA reversed the elution order of the four peptides studied. These investigators claim that "in the literature, PFPA and HFBA generally have been employed in a concentration range of 0.05%–0.1% for the majority of peptide separations" (for HFBA, this is equivalent to 3.8–7.7 mM). Varying the reagent had no advantage for overall separation of peptides with the same net positive charge, but had a profound effect on peptide peak shape and, thus, on resolution.
We have found in the literature that 4–8 mM HFBA indeed has been used for peptides generally. Protein analyses have used 3 mM (79), 15 mM (76,78), and 8–31 mM HFBA (2). In addition, HFBA has been used for: selenium compounds — approximately 8 mM; aminoglycosides, 8–12 mM; herbicides — approximately 15 mM, and most other analytes, 4–8 mM; with a few exceptions below 4 mM (82,94,111) ( B vitamins and epibatidine), and above 8 mM (6,37,48,89,98) (synephrine, norepinephrine, cephalosporin, acetylcholine, and glutamine).
PFPA
The use of PFPA has been sporadic until recent years. Since 1998, it was reported once in 2000, twice in 2002, once each in 2003 and 2005, and then twice, three times, and once in 2006, 2007, and 2008, respectively. PFPA was selected a total of six times for aminoglycosides, its most frequent application.
Debremacker and colleagues (19) noted variation of PFPA from 34–37 mM had only a small positive effect on the separation of a critical pair of spectinomycin anomers and impurities, whereas the pH of the mobile phase, the column temperature, and different C18 columns had larger effects on the separation. Manyanga and colleagues (16,18) separated the pair, gentamicin C2–C2b, with 0.7% TFA and 2.4 mM PFPA (pH 2.6) on a C18 column at 35 °C. They found this separation was influenced mainly by TFA, and the resolution increased when the amount of TFA increased, but decreased with an increase in temperature. Cherlet and colleagues (42,43), and Lecaroz and colleagues (41) both used 20 mM PFPA for the analysis of aminoglycosides. Both de Baere and colleagues (46) (amoxicillin) and Rinne and colleagues (49) (catecholamines) used 9.5 mM PFPA. Chen and colleagues (100) analyzed amodiaquine using 6.3 mM PFPA.
Part II of this article series will continue the discussion of perfluorocarboxylic acids as ion-pair reagents in LC.
References
(1) J.W. Dolan, LCGC 26(2), 170–174 (2008).
(2) S. Delgado, M.C. Garcia, M.L. Marina, and M. Torre, J. Sep. Sci. 26, 1363–1375 (2003).
(3) S. Wybraniec and Y. Mizrahi, J. Chromatogr., A 1029(1–2), 97–101 (2004).
(4) M. Shibue, C.T. Mant, and R.S. Hodges, J. Chromatogr., A 1080(1), 58–67 (2005).
(5) M. Shibue, C.T. Mant, and R.S. Hodges, J. Chromatogr., A 1080(1), 68–75 (2005).
(6) L. Ding, X. Luo, F. Tang, J. Yuan, Q. Liu, and S. Yao, J. Chromatogr., B 857, 202–209 (2007).
(7) C.T. Mant, and R.S. Hodges, J. Chromatogr., A 1125, 211–219 (2006).
(8) D. Slowik-Zilka, K. Safronow, V. Dziedziejko, H. Bukowska, K. Ciechanowski, and D. Chlubek, J. Biochem. Biophys. Meth. 61(3), 313–329 (2004).
(9) B.G. Keevil, S.J. Lockhart, and D.P. Cooper, J. Chromatogr., B 794, 329–335 (2003).
(10) I.T. James, and D. Perrett, J. Chromatogr., A 798, 159–166 (1998).
(11) B. Meddah, S. Kamel, C. Giroud, and M. Brazier, Prep. Biochem. Biotechnol. 29(1), 63–75 (1999).
(12) Y. Shoji, M. Yotsu-Yamashita, T. Miyazawa, and T. Yasumoto, Anal. Biochem. 290, 10-17 (2001).
(13) T.G. Venkateshwaran, J.T. Stewart, R.T. Bishop, J.A. de Haseth, M.G. Bartlett, J. Pharm. Biomed. Anal. 17, 57–67 (1998).
(14) M. Koitka, J. Hochel, D. Obst, A. Rottmann, H. Gieschen, and H. Borchert, Anal. Biochem. 381, 113–122 (2008).
(15) L. Xi, G. Wu, and Y. Zhu, J. Chromatogr., A 1115(1–2), 202–207 (2006).
(16) V. Manyanga, K. Kreft, B. Divjak, J. Hoogmartens, and E. Adams, J. Chromatogr., A 1189, 347–354 (2008).
(17) K. Petritis, P. Chaimbault, C. Elfakir, and M. Dreux, J. Sep. Sci. 24, 397–405 (2001).
(18) V. Manyanga, O. Grishina, Z. Yun, J. Hoogmartens, and E. Adams, J. Pharm. Biomed. Anal. 45, 257–262 (2007).
(19) D. Debremacker, E. Adams, E. Nadal, B. Van Hove, E. Roets, and J. Hoogmartens, J. Chromatogr. A 953, 123–132 (2002).
(20) B. Gammelgaard, L. Bendahl, U. Sidenius, and O. Jons, J. Anal. At. Spectrom. 17, 570–575 (2002).
(21) M. Kotrebai, J.F. Tyson, E. Block, and P.C. Uden, J. Chromatogr., A 866, 51–63 (2000).
(22) M. Kotrebai, M. Birringer, J.F. Tyson, E. Block, and P.C. Uden, Analyst 125, 71–78 (2000).
(23) V. Bonetto, H. Jornvall, M. Anderson, S. Renlund, V. Mutt, and R. Sillard, Eur. J. Biochem. 264, 336–340 (1999).
(24) P. Liu, C.L. Feasley, and F.E. Regnier, J. Chromatogr., A 1047(2), 221–228 (2004).
(25) K.N. Petritis, P. Chaimbault, C. Elfakir, and M. Dreux, J. Chromatogr., A 833, 147–155 (1999).
(26) Z. Spacil, J. Folbrova, N. Megoulas, P. Solich, and M. Koupparis, Anal. Chim. Acta 583(2), 239–245 (2007).
(27) K. Petritis, H. Dessans, C. Elfakir, and M. Dreux, LCGC Eur. February, 98-102 (2002).
(28) K.N. Petritis, P. Chaimbault, C. Elfakir, and M. Dreux, J. Chromatogr., A 833, 147–155 (1999).
(29) N.C. Megoulas and M.A. Koupparis, Anal. Chim. Acta 547(1), 64–72 (2005).
(30) E.G. Galanakis, N.C. Megoulas, P. Solich, and M.A. Koupparis, J. Pharm. Biomed. Anal. 40(5), 1114–1120 (2006).
(31) W.O. Aruda, S. Walfish, and I.S. Krull, LCGC 26(10), 1032–1042 (2008).
(32) K. Risha, J. Flaherty, R. Wille, W. Buck, F. Morandi, and T. Isemura, Anal. Chem. 77, 1503–1508 (2005).
(33) R. Dillert, D. Bahnemann, and H. Hidaka, Chemosphere 67(4), 785–792 (2007).
(34) B.S. Larsen, and M. A. Kaiser, Anal. Chem. 79(11), 3966–3973 (2007).
(35) A.A. Jensen, and H. Leffers, Int. J. Androl. 31, 161–169 (2008).
(36) M.P. Balogh, LCGC 26(6), 540–548 (2008).
(37) J. Keever, R.D. Voyksner, and K.L. Tyczkowska, J. Chromatogr., A 794(1-2), 57–62 (1998).
(38) D. Loffler, and T.A. Ternes, J. Chromatogr., A 1000(1–2), 583–588 (2003).
(39) D.G. Mascher, C.P. Unger, and H.J. Mascher, J. Pharm. Biomed. Anal. 43(2), 691–700 (2007).
(40) C. Lu, and C. Feng, J. Chromatogr., A, 1156, 249–253 (2007).
(41) C. Lecaroz, M.A. Campanero, C. Gamazo, and M.J. Blanco-Prieto, J. Antimicrob. Chemo. 58, 557–563 (2006).
(42) M. Cherlet, S. De Baere, and P. De Backer, J. Mass Spectrom. 35, 1342–1350 (2000).
(43) M. Cherlet, S. De Baere, and P. De Backer, J. Mass Spectrom. 42, 647–656 (2007).
(44) D.N. Heller, J.O. Peggins, C.B. Nochetto, M.L. Smith, O.A. Chiesa, and K. Moulton, J. Chromatogr., B 821, 22–30 (2005).
(45) C. Lu, and C. Feng, J. Sep. Sci. 29, 2143–2148 (2006).
(46) S. De Baere, M. Cherlet, K. Baert, and P. De Backer, Anal. Chem. 74, 1393–1401 (2002).
(47) M.C. Carson and D.N. Heller, J. Chromatogr., B 718, 95–102 (1998).
(48) T.A. Neubecker, M.A. Coombs, M. Quijano, T.P. O'Neill, C.A. Cruze, and R.L.M. Dobson, J. Chromatogr., B 718(2), 225–233 (1998).
(49) S. Rinne, A. Holm, E. Lundanes, and T. Greibrokk, J. Chromatogr., A 1119, 285–293 (2006).
(50) T. Hasegawa, K. Wada, E. Hiyama, and T. Masujima, Anal. Bioanal. Chem. 385, 814–820 (2006).
(51) Y. Zhu, P.S.H. Wong, M. Cregor, J.F. Gitzen, L.A. Coury, and P.T. Kissinger, Rapid Commun. Mass Spectrom. 14, 1695–1700 (2000).
(52) R. Castro, E. Moyano, and M.T. Galceran, J. Chromatogr., A 830(1), 145–154 (1999).
(53) R. Castro, E. Moyano, and M.T. Galceran, J. Chromatogr., A 869(1–2), 441–449 (2000).
(54) J.L.M. Vidal, A.B. Vega, F.J.S. Lopez, and A.G. Frenich, J. Chromatogr., A 1050(2), 179–184 (2004).
(55) M.M. Ariffin, and R.A. Anderson, J. Chromatogr., B 842, 91–97 (2006).
(56) R. Castro, E. Moyano, and M.T. Galceran, J. AOAC Int. 84(6), 1903–1907 (2001).
(57) P. Chaimbault, K. Petritis, C. Elfakir, and M. Dreux, J. Chromatogr., A 855, 191–202 (1999).
(58) P. Chaimbault, K. Petritis, C. Elfakir, and M. Dreux, J. Chromatogr., A 870, 245–254 (2000).
(59) J. Qu, W. Chen, Y. Wang, S. Xiao, Z. Ling, and G. Chen, Analyst 127, 66–69 (2002).
(60) J. Qu, Y. Wang, G. Luo, Z. Wu, and C. Yang, Anal. Chem. 74, 2034–2040 (2002).
(61) T. Teerlink, R. Barto, H.J. ten Brink, and C.G. Schalkwijk, Clinical Chemistry 50, 1222–1228 (2004).
(62) M. Piraud, C. Vianey-Saban, K. Petritis, C. Elfakir, J. Steghens, and D. Bouchu, Rapid Commun. Mass Spectrom.19, 1587–1602 (2005).
(63) A. Tholey, H. Toll, and C.G. Hubel, Anal. Chem. 77, 4618–4625 (2005).
(64) M. de Person, P. Chaimbault, and C. Elfakir, J. Mass Spectrom. 43, 204–215 (2008).
(65) M. Piraud, C. Vianey-Saban, K. Petritis, C. Elfakir, J. Steghens, A. Morla, and D. Bouchu, Rapid Commun. Mass Spectrom. 17, 1297–1311 (2003).
(66) D. Liu, L.W. Beegle, and I. Kanik, Astrobiology 8(2), 229–241 (2008).
(67) M. Piraud, C. Vianey-Saban, C. Bourdin, C. Acquaviva-Bourdain, S. Boyer, C. Elfakir, and D. Bouchu, Rapid Commun. Mass Spectrom. 19, 3287–3297 (2005).
(68) J. Kwon, and M. Moini, J. Am. Soc. Mass Spectrom. 12, 117–122 (2001).
(69) L. Gu, A.D. Jones, and R.L. Last, Anal. Chem. 79, 8067–8075 (2007).
(70) W. Walcher, H. Toll, A. Ingendoh, and C.G. Huber, J. Chromatogr., A 1053(1–2), 107–117 (2004).
(71) K. Petritis, S. Brussaux, S. Guenu, C. Elfakir, and M. Dreux, J. Chromatogr., A 957(2), 173–185 (2002).
(72) M. Orioli, G. Aldini, G. Beretta, R.M. Facino, and M. Carini, J. Chromatogr., B 827(1), 109–118 (2005).
(73) Y. Ishihama, H. Katayama, and N. Asakawa, Anal. Biocem. 287, 45–54 (2000).
(74) M. de Person, A. Sevestre, P. Chaimbault, L. Perrot, F. Duchiron, C. Elfakir, Anal. Chim. Acta 520, 149–158 (2004).
(75) M. Orioli, G. Aldini, M.C. Benfatto, R.M. Facino, and M. Carini, Anal. Chem. 79, 9174–9184 (2007).
(76) M.C. Garcia, A.C. Hogenboom, H. Zappey, and H. Irth, J. Chromatogr., A 957(2), 187–199 (2002).
(77) S.A. Gustavsson, J. Samskog, K.E. Markides, and B. Langstrom, J. Chromatogr., A 937, 41–47 (2001).
(78) Y. Wang, B.M. Balgley, P.A. Rudnick, and C.S. Lee, J. Chromatogr., A 1073(1-2), 35–41 (1–2).
(79) M. Naldi, V. Andrisano, J. Fiori, N. Calonghi, E. Pagnotta, C. Parolin, G. Pieraccini, and L. Masotti, J. Chromatogr. A 1129, 73-81 (2006).
(80) R.M. Kok, D.E.C. Smith, R. Barto, A.M.W. Spijkerman, T. Teerlink, H.J. Gellekink, C. Jakobs, and Y.M. Smulders, Clin. Chem. Lab. Med. 47(7), 903–911 (2007).
(81) T.M. Dillon, P.V. Bondarenko, and M.S. Ricci, J. Chromatogr., A 1053(1–2), 299–305 (2004).
(82) A.P. Watt, L. Hitzel, D. Morrison, and K,L, Locker, J. Chromatogr., A 896(1–2), 229–238 (2000).
(83) K. Petritis, V. Dourtoglou, C. Elfakir, and M. Dreux, J. Chromatogr., A 896(1–2), 335–341 (2000).
(84) M.R. Hakkinen, T.A. Keinanen, J. Vepsalainen, A.R. Khomutov, L. Alhonen, J. Janne, and S. Auriola, J. Pharm. Biomed. Anal. 45, 625–634 (2007).
(85) L. Vernez, G. Hopfgartner, M. Wenk, and S. Krahenbuhl, J. Chromatogr. A 984(2), 203–213 (2003).
(86) D. Sun, M.G. Cree, X. Zhang, E. Boersheim, and R.W. Wolfe, J. Lipid Res. 47, 431-439 (2006).
(87) D.E. Starkey, J.E. Denison, C.T. Seipelt, and W.A. Jacobs, J. AOAC Int. 91(1), 130–142 (2008).
(88) P. Andrieux, T. Kilinc, C. Perrin, and E. Campos-Gimenez, J. AOAC Int. 91(4), 777–785 (2008).
(89) P. McCormack, P.J. Worsfold, and M. Gledhill, Anal. Chem. 75, 2647–2652 (2003).
(90) T.D. Grant, M. Montes-Bayon, D. LeDuc, M.W. Fricke, N. Terry, and J.A. Caruso, J. Chromatogr., A 1026(1–2), 159–166 (2004).
(91) T. Lindemann, and H. Hintelmann, Anal. Bioanal. Chem. 372, 486–490 (2002).
(92) X. Fang, X. Fan, Y. Tang, J. Chen, and J. Lu, J. Chromatogr., A 1036(2), 233–237 (2004).
(93) T. Hashimoto, S. Nishio, N. Nishibori, S. Yoshioka, and T. Noguchi, J. Food Hyg. Soc. Japan 43(3), 144–147.
(94) O. Midttun, S. Hustad, E. Solheim, J. Schneede, and P.M. Ueland, Clinical Chemistry 51, 1206–1216 (2005).
(95) S. Gao, S. Bhoopathy, Z. Zhang, D.S. Wright, R. Jenkins, and H.T. Karnes, J. Pharm. Biomed. Anal. 40(3), 679–688 (2006).
(96) J.X. Shen, E.A. Merka, D.P. Dreyer, R.P. Clement, and R.N. Hayes, J. Sep. Sci. 31, 242–254 (2008).
(97) J. Lu, Y. Chan, S.D. Poppitt, and G.J.S. Cooper, J. Chromatogr., B 859(1), 62–68 (2007).
(98) J.A. Eckstein, G.M. Ammerman, J.M. Reveles, and B.L. Ackerman, J. Neurosci. Methods 171, 190–196 (2008).
(99) Y. Aoki, H. Hakamata, Y. Igarashi, K. Uchida, H. Kobayashi, N. Hirayama, A. Kotani, and F. Kusu, J. Chromatogr., B 858, 135-142 (2007).
(100) X. Chen, P. Deng, X. Dai, and D. Zhong, J. Chromatogr., B 860, 18–25 (2007).
(101) N. Kaga, S. Soma, T. Fujimura, K. Seyama, Y. Fukuchi, and K. Murayama, Anal. Biochem. 318, 25–29 (2003).
(102) F. Jin, J. Hu, M. Yang, X. Jin, W. He, and H. Han, J. Chromatogr., A, 1101, 222–225 (2006).
(103) A.O. De Silva and S.A. Mabury, Environ. Sci. Technol. 38, 6538–6545 (2004).
(104) K. Petritis, C. Elfakir, and M. Dreux, LCGC Eur. 14(7), 389–394 (2001).
(105) P. Petersson, M. Jorten-Karlsson, and M. Stalebro, Electrophoresis 24, 999–1007 (2003).
(106) M. Tsuji, T. Inoue, and O. Shibata, Colloids Surfaces B: Biointerfaces 61, 61–65 (2008).
(107) D.N. Heller, S.B. Clark, and H.F. Righter, J. Mass Spectrom. 35, 39–49 (2000).
(108) Z. Spacil, J. Folbrova, N. Megoulas, P. Solich, and M. Koupparis, Anal. Chim. Acta 583(2), 239–245 (2007).
(109) K.A.J. Snoble, N.M. Fox, S.M. Lorenz, A.R. Kemperman, and J.T. Przybytek, LCGC 26(9), 946–950 (2008).
(110) K. Petritis, P. Chaimbault, C. Elfakir, and M. Dreux, J. Chromatogr., A 896(1–2), 253–263 (2000).
(111) O. Midttun, S. Hustad, E. Solheim, J. Schneede, and P.M. Ueland, Clin. Chem. 51(7), 1206–1216 (2005).


















New Study Reviews Chromatography Methods for Flavonoid Analysis
April 21st 2025Flavonoids are widely used metabolites that carry out various functions in different industries, such as food and cosmetics. Detecting, separating, and quantifying them in fruit species can be a complicated process.



