Removal of Contaminant Peaks in Reversed-Phase Gradient Liquid Chromatography for Improved Detection of Pharmaceutical Impurities
LCGC Asia Pacific
This article presents a method for comparing the levels of baseline interference arising from common laboratory mobile phase contamination sources and assesses different approaches for removing dissolved contaminants to generate interference-free chromatogram baselines. The authors demonstrate that recirculating mobile phase through a semi-preparative scale column using a reagent delivery pump has advantages over previously published mobile phase decontamination methods.
Mark R. Taylor1, Fiona Harvey-Doyle1, and Krina Patel2,3
1Dept. Analytical Research and Development, Pfizer Worldwide Research and Development, Sandwich, Kent, UK,
2Dept. Forensic Science & Drug Monitoring, King’s College London, London, UK,
3Present address: Dept. of Chemistry, University of Southampton, Southampton, UK.


Image credit: MakiEni's photo
This article presents a method for comparing the levels of baseline interference arising from common laboratory mobile phase contamination sources and assesses different approaches for removing dissolved contaminants to generate interference-free chromatogram baselines. The authors demonstrate that recirculating mobile phase through a semi-preparative scale column using a reagent delivery pump has advantages over previously published mobile phase decontamination methods.


Increasing regulatory scrutiny combined with improved modern liquid chromatographs used in pharmaceutical purity analysis has seen a trend towards increased detection sensitivity. It is now commonplace in pharmaceutical analysis to measure impurities and degradants at sub-nanogram concentrations from sample column loads of 200 nanograms or less when using UV detection. When performing such high-sensitivity measurements, complications with background environmental contamination of the system and mobile phase are often encountered. Commonly referred to as ghost peaks but more properly denoted as contaminant peaks, extraneous signals arising from UV absorbing contaminants in the liquid chromatography (LC) system can interfere with measurements of low-level solutes. Unless special measures are taken the interference from contaminant peaks can lead to inaccurate measurements of low-level impurities.
Modern chromatographs with low dead volumes and high sensitivity UV detectors are commonly used in pharmaceutical analysis (1,2,3) with gradient elution to separate diverse polarity solute mixtures of active pharmaceutical ingredients (APIs) and impurities within reasonable run times (4). UV detection is still commonly preferred to mass spectrometry (MS) because of reduced cost and ease of methods transfer. Laboratories are often required to quantify solutes at sub-nanogram concentrations (5) when measuring low levels of potentially genotoxic impurities (6) and testing the effectiveness of cleaning methods (7). According to current industry guidelines (8), methods for the purity analysis of pharmaceuticals should be capable of quantifying impurities above a 0.03% threshold for drugs with a maximum dose of more than 2 g/day. For doses of less than 2 g/day the threshold is relaxed to 0.05% of API concentration. In early development stages reference standards are often unavailable for all solutes of interest and a peak area normalization approach is commonly used to monitor and control impurity levels. When working at such extremes of sensitivity in industrial environments it can be difficult to avoid interference from environmental contamination unless special measures are taken.
In modern industrial settings detection of unwanted extraneous contaminant peaks in the chromatogram baseline can interfere with measurements (9,10) and require additional time and effort to identify the cause and implement counter-measures. These can have a significant impact on the transferability of analytical methods because of differences in the purity of reagents and handling practices at each site - often the causes remain unresolved (11,12). Extraneous peaks in the chromatogram that are not directly related to the injected sample of interest have been referred to as system peaks (13), vacancy peaks (14), artefact peaks (15), and induced peaks (16). However, the most commonly used term is “ghost peaks” (17,18), and because they usually arise from some kind of system contamination it is more correct to refer to them as “contaminant peaks” (CP).
Since the advent of gradient liquid chromatography (LC) baseline interferences arising from mobile phase contamination has been a problem and many publications have been devoted to this subject (19,20,21,22,23,24). Gradient elution exacerbates contamination problems because trace contaminants become enriched on the column and then elute once the elutropic strength becomes sufficiently high. There are lots of possible sources of contamination in the modern LC laboratory and trace contamination can be very difficult to avoid. Contamination of water with airborne particles, organics, colloids, and ions (11,25), impure additives (19-24,26), dissolved gases, and organic solvent impurities (11,27,28) have all been reported, as has accidental contamination from pH meters and laboratory glassware (19-24).
It has been recommended that LC mobile phases should be freshly prepared and micro-filtered before use (29) and that LC solvent lines and glassware be regularly rinsed to avoid build-up of biofilms and detergents (30) and because of the propensity for microbial growth in buffers. Plasticizers such as phthalates are prevalent in the environment where indoor air levels have been detected above 10 µg (31). Phthalates can be detected on most surfaces and are readily extracted by organic solvents (32). In a laboratory setting plasticizers were detected in laboratory water and leaching from consumables (33). Adsorption of plasticizers from the air can compromise the long-term stability of reagents (34) and the concentrations of these contaminants can be increased by exposure to laboratory equipment and manual handling steps (35).
To remove contaminant interference in LC various methods for contaminant extraction from the mobile phase have been published. Solid-phase extraction (SPE) using hydrophobic sorbents has been applied with some success (36,37,38). However SPE under vacuum is prone to flow-channelling and breakthrough of more polar impurities, particularly with membrane disks, which have limited retention capacity as a result of short bed length (39). Variability in contaminant removal effectiveness has also been reported (40,41).
Mobile phase decontamination using in-line guard columns helps to remove micro-particulates and prolongs the lifetime of analytical columns. However, because they remain in-line to the analytical column, they are not a reliable way of removing contaminant peaks. The use of high performance liquid chromatography (HPLC) columns as impurity traps (installed between pump and mixer) has been shown to be effective at removing dissolved trace contaminants from the aqueous phase, resulting in reduced levels of baseline interference, but the use of short trap columns limits the effective lifetime of the trap before contaminant breakthrough occurs (42). This is particularly true for more polar contaminants, which can break through the trap columns and refocus on longer analytical columns to cause interference.
The use of longer, larger capacity trap columns can be limited by the undesired contribution to extra system volume if applied directly to the analytical LC pump. To do this efficiently the trap column needs to be of sufficient scale to retain contaminants from a large number of gradient cycles and operate throughout a run sequence. Using a reagent delivery pump it is possible to perform mobile phase decontamination outside of the flow-path of the analytical LC system. In this novel configuration the trap column does not contribute to system back-pressure or dwell volume and can be applied with any type of gradient LC pump. On-line continuous clean-up of mobile phase using longer bed length and larger capacity trap columns protects against subsequent airborne contamination without the need to flush the trap between injections.
This article presents a method for comparing the levels of a LC-UV chromatogram baseline interference arising from mobile phases exposed to different contaminants and decontamination methods in an attempt to highlight the highest risk sources of mobile-phase contaminant peak generation and test the effectiveness of various approaches for decontamination of the mobile phase.
Experimental
Mobile Phase Preparation: The (95% v/v) aqueous mobile phases were prepared using ammonium acetate (HPLC grade, Thermo Fisher Scientific) to a concentration of 10 mM with addition of acetonitrile (5% v/v, Sigma-Aldrich). Purified water was obtained from a Milli-Q Gradient A10 Purification system (Merck Millipore). Mobile phase solutions were stored at ambient laboratory conditions during the evaluation. Larger batches were stored in 10 L polypropylene bottles and smaller sub-lots used in systematic tests were stored in 1 L capacity clear glass Duran bottles that were washed thoroughly with organic solvent (acetonitrile) and allowed to dry (inverted) before use.
Evaluation of Contamination Sources: To determine the sources of potential contamination, mobile phases were deliberately contaminated by objects commonly found in the laboratory: a plastic weighing boat, dishwasher detergent, Tygon tubing, Parafilm, contaminated pipette, and two different types of nitrile glove; the thin single-use disposable type often used for general laboratory tasks and the thicker reusable type. Dust particles were collected from our own laboratory windowsill. A 5-L batch of ammonium acetate buffer (native pH measured at 6.8) containing 5% acetonitrile was prepared and 500-mL lots were subdivided into clean glass bottles, which were rinsed out before use with high-quality acetonitrile. Each bottle was deliberately contaminated by immersing solid items (for example, gloves, Parafilm) in the mobile phase for 20 s. A small pinch of dust (c.a. 0.5 mg) was sprinkled onto the surface of one bottle for comparison. Each contaminated mobile phase lot was used to generate a baseline following enrichment of 60 mL of the mobile phase onto the column before running an acetonitrile gradient program to elute the contaminants. To test the efficacy of the clean-up techniques under worst-case conditions, a 5-L stock solution was prepared and then sub-aliquotted into 500-mL lots, each of which was deliberately contaminated with the common laboratory products mentioned previously. The mobile phase was tested before and after each of the different clean-up techniques and compared to an unadulterated batch of the stock solution.
Mobile Phase Diluents: To test the purity levels of water and acetonitrile a number of different grades and sources were obtained to test any differences between suppliers and grades. Water was also obtained from a Milli-Q Gradient A10 purification system (Merck Millipore).
Off-Line Extraction Techniques: Emporestyrenedivinylbenzene (SDB-XC) 47-mm SPE filter disks were obtained from Supelco. Oasis Hydrophilic Lipophilic Balance (HLB) (6 g) packed particle SPE cartridges were obtained from Waters, and Envi-Carb 12-mL (1 g) and 3-mL (0.25 g) porous-graphitic carbon (PGC) SPE cartridges from Sigma Aldrich. For the off-line SPE techniques, a vacuum manifold (Waters) was used. The adulterated mobile phase (500 mL) was passed through each SPE cartridge following pre-conditioning with methanol (6 mL) and purified water (6 mL) and collected in a glass bottle pre-rinsed with acetonitrile. For the filter disks, a glass vacuum filtration apparatus (Millipore) pre-cleaned with acetonitrile was used to perform the filtration. Each SPE device was pre-washed with acetonitrile (10 mL) and purified water (10 mL) before passing 500 mL of mobile phase through under slight vacuum. Stir-bar sorptive extraction (SBSE) stir-bars (4 cm, coated with polydimethylsiloxane (PDMS) were donated by RiChrom. The extraction was performed by stirring 500 mL of aqueous mobile phase at a medium speed (c.a. 120 rpm) for 60 min before sampling. All treatments were prepared in singlicate.
In-Line and On-Line Extraction Techniques: A Turboflow Cyclone-MCX column (4.6 × 100 mm, 50-µm) containing a styrene divinylbenzene (SDB) copolymer and a Turboflow Silica-C18 column (4.6 × 100 mm, 50-µm) were donated by Cohesive Technologies. Ghost-trap DS micro-columns (4.0 × 20 mm) were purchased from Shimadzu. Luna A C18 (2) semi-preparative column (150 × 10 mm, 5-µm and 150 × 21.2 mm, 5-µm) were obtained from Phenomenex. The trap columns were evaluated by placing them (a) in-line between pump-head and mixer (for turbulent flow chromatography (TFC) columns only) and (b) connecting the C18 columns on-line by pumping into the mobile phase inlet line using an additional model 422 isocratic pump (2 mL/min, Kontron) via a tee-piece with an overflow line back to the reservoir.
Oxycodone (OC, Sigma Aldrich) standard testing was performed using Agilent 1100 series HPLC systems comprising of an on-line degasser, low pressure mixing quaternary pump with proportioning valve, auto-sampler, and diode array UV detector with Empower software control (Waters Inc.). UV detection wavelength was 210 nm. Standards were prepared at concentrations of 200 µg/mL and 100 ng/mL and 10 µL injections were made onto a 4.6 × 150 mm, 3.5-µm XBridge C18 column (Waters Inc.) at a flow rate of 1 mL/min. Mobile phase was ammonium hydroxide (Fluka LC-MS grade 1 mL phials, Sigma Aldrich, diluted to a final concentration of 0.1% v/v with purified water) and acetonitrile. A gradient elution programme was performed by changing the acetonitrile composition from 5% to 64% over 29 min following a 2.5 min hold at 5% acetonitrile with a flow-rate of 1 mL/min. Mobile phase composition was maintained at 64% acetonitrile until 34.5 min, after which the column was re-equilibrated at 5% acetonitrile for 5 min. Column temperature was maintained at 32 °C, which was previously determined as the optimum for the separation of OC and its impurities during method development activities.
Instrumentation: HPLC separations investigating the sources of mobile phase contamination and the effectiveness of removal methods were performed using two different Agilent 1200 series HPLC systems comprising of an on-line degasser, binary pump with high-pressure static mixer, autosampler, and diode array UV detector with Chemstation software control (Agilent Technologies Inc.). UV detection wavelengths of 210, 240, and 254 nm were used for detection. Chromolith C18 monolithic HPLC columns (10 × 4.6 mm, obtained from Merck) were used for the trapping, elution, and resolution of enriched contaminants. To study the sources of contamination, zero volume injections were performed following equilibration of the column for 20 min at a flow-rate of 3 mL/min and organic composition of 5% acetonitrile. Following equilibration a gradient ramp programme of 5-95% acetonitrile over 10 min was used to elute the enriched contaminants from the column. For the evaluation of clean-up techniques, the equilibration time was reduced to 5 minutes prior to running a gradient of 5-95% acetonitrile over 10 min. Duplicate measurements were made for each tested lot of mobile phase. The first measurement was discarded and the second replicate was used for the comparison. To avoid cross-contamination systems were washed by flushing lines with 80:20 (v/v) acetonitrile:water before use. Gradient-grade (≥99.9%) acetonitrile (Sigma Aldrich) was used throughout the clean-up and contamination experiments. Trap columns of all sizes were prepared by flushing with MS-grade acetonitrile for 15 min followed by water (obtained from the MilliQ system) containing 5% acetonitrile for 15 min at 1 mL/min (micro- and analytical-scale traps) and 10 mL/min (semi-prep and preparative column traps).
Results

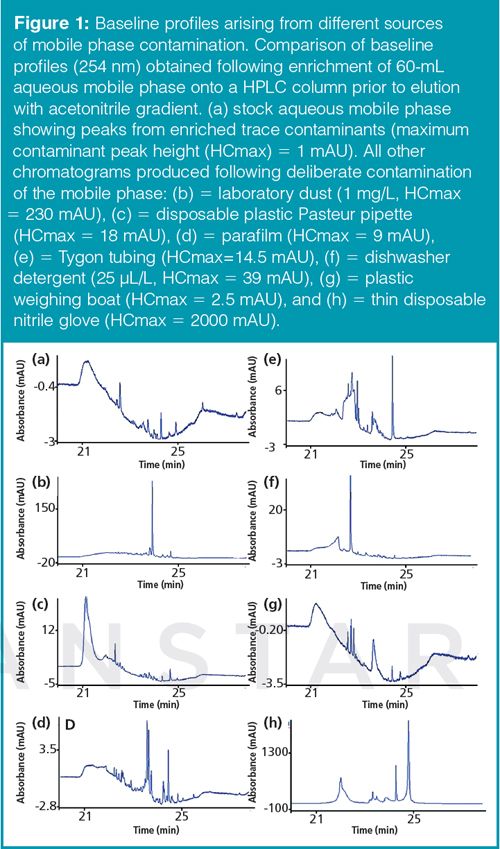
To be able to measure and compare the level of contamination arising from different mobile phase treatments, a method was developed to equilibrate a column with 60 mL mobile phase under highly aqueous conditions before eluting contaminants using a gradient elution cycle. Baselines were monitored at a range of wavelengths typically used in pharmaceutical analysis by LC-UV. Comparison of different contamination sources by exposing mobile phase to different contaminants confirmed that common laboratory equipment could have a significant impact on the formation of contaminant peaks. Example chromatograms from these studies are provided in Figure 1. All tests showed some degree of contamination would accumulate if more than one contamination source was exposed to the mobile phase during preparation.
Disposable nitrile gloves are frequently used in many LC laboratories. Though it would not be considered normal practice to immerse a whole glove into a mobile phase container, it is common to handle mobile phase transfer lines, filter sinkers, bottle necks, and caps whilst wearing these gloves, and the high level of mobile phase contamination observed from this source indicate that they are a potential risk for generation of unwanted contaminant peaks. Dust contamination of the mobile phase also generated a significant increase in the contaminant peak profile. Dust is always present in laboratory air as a result of human activity and accidental contamination of mobile phase lines, filter sinkers, and bottles by leaving them exposed to the air is also considered to be a high risk activity and can lead to contaminant peak interferences.

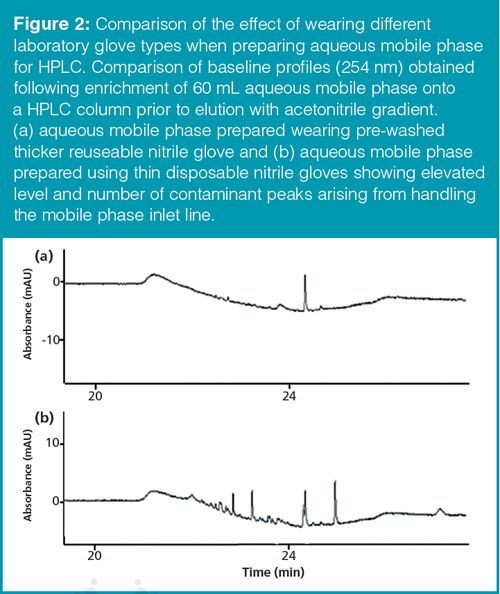
To mimic normal mobile-phase handling procedures, separate lots of mobile phase were prepared whilst wearing different types of glove. These included weighing and dilution of the salt, rinsing and filling of glass mobile phase bottles, and transferring the (pre-washed) solvent lines between mobile phase bottles. Results (Figure 2[a]) showed minimal additional contamination arising from use of (pre-washed) nitrile gloves compared to wearing the softer disposable nitrile gloves (Figure 2[b]), where significant extra contaminant peaks were observed, though in a much lower scale to when the whole glove was immersed. Leachable plasticizers from disposable gloves are likely to also be deposited on the hands and be transferred to anything touched after removal, including LC solvent lines and fittings, and as a result of this study the authors recommend that they should not be worn when handling anything in the LC flow path.
Other sources of plasticizers such as plastic Pasteur pipettes, Parafilm, plastic tubing, and plastic weighing boats also gave rise to a significant increase in the number and level of contaminant peaks but at lower levels to those observed from gloves and dust. During this study it became evident that the size of some contaminant peaks present in the reference blank stock mobile phase solution increased significantly over time, presumably as a result of airborne contamination, microbial growth, or leaching from container, LC inlet filter sinker, solvent lines, or cap.
Experiments were performed to determine and compare the baseline quality arising from a range of different grades of water and acetonitrile using the same method as described previously. It was observed that mobile phase prepared with commercial bottled HPLC gradient grade water produced lower quality baselines than mobile phase-prepared water from the laboratory water purification system. The apparent lower quality of commercial bottled water was consistent with previously published results (22). This could possibly be a result of the manufacturer’s purification system, insufficient container cleaning, cross-contamination with other solvents, microbes, environmental contamination, and leachates from storage vessels and caps. Laboratory water purification systems are capable of producing water with 5-50 ppb organic content, but only if they are regularly maintained and flushed. In line with previously published reports (24, 25) we also observed effects on baseline quality from different grades of acetonitrile. The baselines showed no significant difference between them in terms of the levels of observed contaminant peaks but the background UV response was higher with acetonitrile from some suppliers than from others. The LC-MS grade acetonitrile used in this study produced a generally lower baseline response compared to cheaper commercial HPLC grades. A possible explanation for this could be down the manufacturing process of acetonitrile because it is produced as a by-product from the large-scale production of acrylonitrile, which possesses a stronger chromphore than acetonitrile. We concluded that the highest available quality acetonitrile should be used when analyzing low-level solutes or when low UV detection wavelengths are required.
Different grades of ammonium acetate buffer salt were also compared. Results showed significant variations in baseline quality arising from the use of each batch. Surprisingly, the largest contaminant peaks were observed from the use of one of the highest-grade (99.999+ label purity) materials investigated. This buffer salt was supplied in a soft plastic screw-cap bottle, so it is possible that plasticizer leachates were responsible for these additional peaks. The level of contamination originating from the container could also possibly be exacerbated by scraping the sides of the container during weighing and by the deliquescent nature of ammonium acetate.
A range of extraction techniques were evaluated for their effectiveness in removing the contaminant peaks from identical lots of mobile phase prepared by deliberately adulterating clean aqueous ammonium acetate (containing 5% acetonitrile) with a range of different contamination sources. This was compared by summing the peak areas of those peaks eluting in the first half of the mobile phase gradient cycle (5-50% acetonitrile) and the second half of the cycle (50-95% acetonitrile) of each chromatogram separately so that any trends related to contaminant polarity could be observed. The degree of change in total contaminant peak response resulting from each treatment was compared (Table 1).
Two commercial SPE cartridges packed with different stationary phases and a hydrophobic membrane filter disk were compared for their effectiveness at the removal of polar and non-polar contaminants from 500 mL of contaminated mobile phase. One cartridge was more effective in reducing the overall level of contaminant peaks but the other was more effective at removing non-polar contaminants (see Table 1). Despite thorough pre-rinsing of the cartridge, some additional signals (max peak height 5 mAU) were observed in the polar contaminant region of the chromatogram baseline after treatment with the latter cartridge possibly arising from leaching of UV absorbing components from stationary phase, frits, or column. Vacuum SPE, though simple to use, required significant additional processing by the operator and was found to be time-consuming and limited to small volumes. Using SPE devices on solvent inlet lines has been evaluated in our laboratories and found to be ineffective because channelling and cavitation affected the consistency of flow and effectiveness of contaminant removal.
The membrane filter disk SPE device was less effective at removing contaminants than the packed-bed SPE devices (also found in previously published results [40]), presumably as a result of reduced solute retention capacity from the reduced sorbent mass and bed depth. The flow-rate through the disk was very slow and the filtration process was time-consuming. This would be a limitation when large volumes need to be prepared. It is possible that repeating measurements using more diverse chemistries and more replicates may give a more accurate picture of the capabilities of off-line clean-up techniques for contaminant removal. However, the large volumes of mobile phase sometimes required and the use of off-line clean-up techniques leaves the mobile phase prone to subsequent contamination following the initial clean from airborne plasticizers, dust, and microbes made this unfavourable.
SBSE was similarly tested for its effectiveness in the removal of contaminants. This method is highly attractive because of its simplicity and the fact that the mobile phase can be continually cleaned by spinning the stir-bar throughout the run time of the LC. Unfortunately for this application the approach was limited by the small stir-bar devices available. Larger capacity SBSE devices would be worthy of development for this particular application, perhaps using porous monoliths, but care would need to be taken to avoid phase-bleed.
On-line continuous clean-up techniques are advantageous because mobile phase purification occurs at or close to the point of use. Trap micro-columns made specifically for removal of contaminant peaks were placed between an aqueous pump and mixer on both HPLC and ultrahigh-pressure LC (UHPLC) systems and some contaminant removal was observed during the first few runs. However, contaminant breakthrough from the trap micro-columns occurred very quickly. As a result of their small size such columns have limited retention capacity and may be unsuitable for long sequence runs without continual flushing and regeneration as previously demonstrated (20, 22-24).
In an attempt to increase the contaminant trapping capacity, the micro-column was replaced with larger scale (4.6 × 100 mm) turbulent flow chromatography (TFC columns). TFC adopts the use of large (typically 50-µm) porous particles allowing for high flow rates with low back pressure in the system (43), though with the flow-rates used in this study (1-3 mL/min) turbulent flow conditions would not be achieved. Two stationary phases were compared and both were found to be more efficient at removing the contaminant peaks than the lower-capacity micro-column trap (Table 1). The bonded silica C18 column was more effective at removing the contaminant peaks than the polymeric column where some small peaks were still observed in the polar region of the chromatogram. Clean up of the traps necessitated removal from the system, which is undesirable because handling mobile phase lines risks further system contamination.
A limitation of siting a trap column between the mixer and the pump is that the trap column will contribute to the overall pump back pressure. Furthermore, this configuration is not possible with low-pressure mixing systems using proportioning valves because the aqueous mobile phase needs to be pumped through the phase to avoid cavitation and flow-channelling. Trying to aspirate the solvent through the trap would result in undesirable cavitation and flow-channelling.

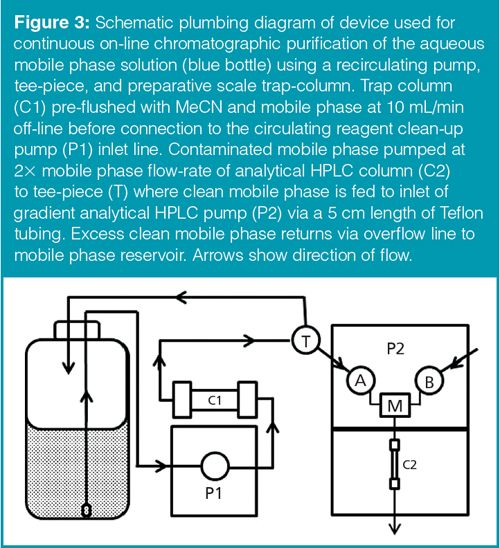
To overcome the additional pressure limitation and system volume increase when using even larger trap columns, a new system was designed to allow the mobile phase to be recirculated through a semi-preparative scale trap column using an additional reagent delivery pump and low-pressure PEEK Tee-piece (Figure 3). Because the trap column was out of the flow-path of the LC column it was possible to use the larger trap columns without adversely affecting the chromatography. A semi-prep scale bonded silica C18 column (10 × 150 mm, 5-µm) was shown to remove 95% of the polar contaminant peaks and 100% of the non-polar contaminant peaks from the grossly contaminated mobile phase throughout 24 h of continuous operation (60 tests) leaving a clean baseline throughout the course of the run.

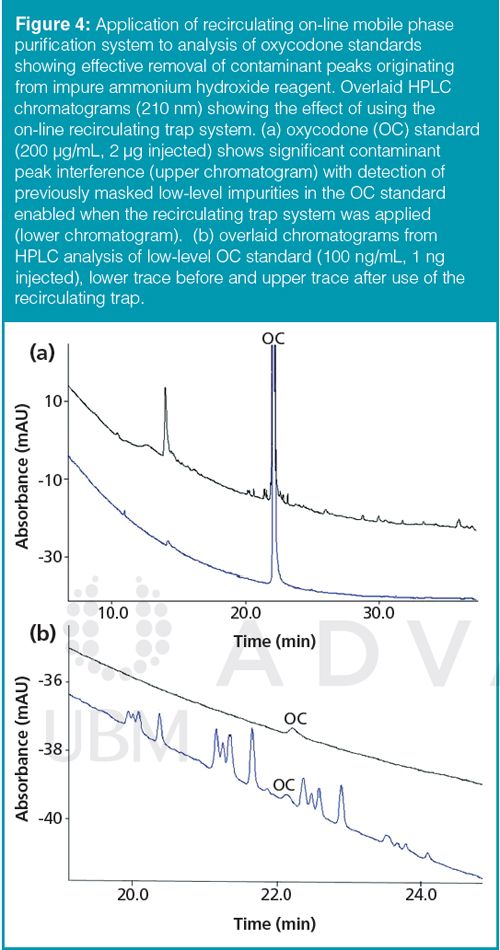
To test the effectiveness of the recirculating mobile phase purification system in a commercially validated method it was applied to the purity testing of pharmaceutical samples containing oxycodone (OC). This method had proved troublesome in transfer between testing sites because of the presence of contaminant peaks originating from ammonium hydroxide and/or water used to prepare the mobile phase. The contaminant peaks interfered with the low level determination of OC and OC impurities by HPLC with UV detection. Contaminant peak profiles were found to vary from site to site and between different lots of reagent from the same supplier. This resulted in time wasted during investigations and the need to repeat analyses with fresh batches of mobile phase. Using a larger trap column the recirculating mobile phase clean-up system was successfully applied to this method. The interfering compounds from the mobile phase were removed and trouble-free operation was achieved throughout the two-week course of the study. Typical results from analysis of standards showing the baseline before and after cleaning are shown in Figure 4. Following completion of the study the trap was flushed with acetonitrile to remove the retained contaminants, thereby removing the need to handle the column or solvent lines.
Discussion
Successful method transfer between laboratories is at risk from contamination events arising from different reagent supplies and qualities and laboratory cleanliness and handling practices. The results obtained from these experiments are comparable to previous studies (17,20,â¨22-26), which showed that common laboratory consumables and the quality of reagents and solvents used in mobile phase preparation can have a significant impact on the formation of additional ghost peaks. Sub-optimal handling practices such as handling mobile phase lines whilst wearing plasticizer-laden gloves exacerbates the problem.
It is generally recommended that aqueous mobile phase should be freshly prepared to avoid microbial contamination of the water and contamination from airborne contaminants. However, it is very difficult to avoid contaminant peaks completely as a number of factors can cause them. The ubiquity of plasticizers and dust in modern laboratories makes it very difficult to completely avoid trace contamination of mobile phase occurring. It is recommended where possible to avoid the use of plastic materials in LC mobile phase and sample preparation particularly for high sensitivity LC-UV methods where solutes need to be measured at low concentrations. Typical laboratory air contains comparatively large quantities of dust and other airborne particles such as semi-volatile organic compounds (SVOCs) from air handling systems, which can readily adhere to surfaces and glassware and cause contamination. Therefore it is necessary to protect solutions and solvent lines from airborne contamination. Detergents used in glassware washing can also contribute to the contaminant peak profile unless residues are thoroughly rinsed from the glassware before use.
The recirculating purification system that used large-scale HPLC columns proved to be the most effective of all the extraction techniques tested, removing almost all of the contaminant peaks for sustained periods of operation without compromise to system pressure or adding additional system volume. The columns were simply regenerated by flushing with acetonitrile after each run sequence or when required. The plumbing configuration using a separate reagent delivery pump is compatible with LC systems using both low and high pressure mixers without any compromise to system dead or dwell volume or pressure limitation as would be observed if the trap column were placed in-line. The mobile phase is constantly cleaned so any contaminants adsorbed from the atmosphere and other sources are continually removed before entering the analytical LC pump. In addition, large volumes of mobile phase can be cleaned through the trap column since the size of the column and stationary phase can be easily increased.
The system does require an additional pump and plumbing, which, although easy to connect to any type of LC gradient pump, may prove prohibitive to routine use in some laboratories on the basis of cost. However, when faced with the need to measure multiple trace sample components without reference standards (as is common for pharmaceutical stability studies) and to transfer methods between sites operating with different quality water, solvent, and reagent supplies, it is more practical and cost-effective than resorting to MS for quantitative analysis. Furthermore, reducing the number of failed runs and investigations and speeding up data processing through routine generation of interference-free baselines easily offsets the extra cost. Application of this approach to cleaning of the organic solvent may enable the use of lower-cost solvent grades, though the effective purification of the aqueous mobile phase has the greatest positive impact on the quality of baselines generated in gradient reversed-phase LC.
Conclusions
Even with ultra-clean handling processes it is very difficult to avoid trace contamination of the mobile phase in modern industrial laboratories. Contaminant peak interference in gradient LC is commonplace and even handling solvent lines whilst wearing gloves contributes to the contaminant peak profile. The identification and resolution of contaminant peaks from target analyte peaks of low-level impurities is time consuming and risks the quality and accuracy of purity measurements made in pharmaceutical release testing and stability studies. In this article a method is presented to eliminate the problem, allowing the operation of gradient LC-UV for extended periods of time. The system was simple to set up and scalable for use with different sizes of column, different column chemistries, and with any type of pump. Column regeneration and conditioning was also simple. Future gradient pump design could potentially incorporate reagent clean-up systems like this to help overcome the baseline problems caused by trace-level contaminants entering the pump and interfering with detection in LC-UV.
Acknowledgements
The authors would like to thank Dr Frank David (RiChrom) for his donation of the SBSE stir-bars and Dr Hubert Quinn (Cohesive Technologies) for donation of the preparative scale TFC columns. Thanks to Dr Melissa Hanna-Brown and Dr Jonathan Beaman (Pfizer) and Dr Mark Parkin (King’s College, London) for their advice and assistance with manuscript review.
References
- S. Chesnut and J. Salisbury, J. Sep. Sci.30, 1183-1190 (2007).
- A. Jerkovich, R. LoBrutto, and R. Vivilecchia, Pharm. Anal. Dev.1, 15-21 (2005).
- S. Cai, J. Stevens, and J. Syage, J. Chromatogr. A.1227, 138-144 (2012).
- S. Das, S. Mohapatra, and J. Hsu, J. Liq. Chromatogr. Relat. Technol.23, 1809-1819 (2000).
- F. Qui and D. Norwood, J. Liq. Chromatogr. Relat. Technol.30, 887-935 (2007).
- A. Giordani, W. Kobel, and H. Gally, Eur. J. Pharm. Sci.43, 1-15 (2011).
- A. Plasz, Sep. Sci. Technol.6, 401-412 (2005).
- International Conference on Harmonization, ICH Q3A (R2), Impurities in New Drug Substances, ICH Harmonised Tripartite Guideline (ICH, Geneva, Switzerland, 1997).
- S. Levin and E. Grushka, Anal. Chem.58, 1602-1607 (1986).
- B. Buszewski, S. Bocian and A. Felinger, Chem. Rev.112, 2629-2641 (2012).
- S. Sadikin, D. Zhang, R. Inloed, and S. Redkar, Pharm. Technol.1, 1-9 (2011).
- Y. Egi and A. Ueyanagi, LCGC North Am.16, 1-4 (1998).
- N. Mizrotsky, L. Kristol, and E. Grushka, J. Chromatogr. A.691, 21-27 (1995).
- C. Desiderio, S. Fanali, and P. Gebauer, J. Chromatogr. A.772, 81-89 (1997).
- G. Strasser and I. Varadi, J. Chromatogr. A.869, 85-90 (2000).
- W. Hammers, C. Aussems, and M. Janssen, J. Chromatogr. A.360, 1-12 (1986).
- S. Williams, J. Chromatogr. A.1052, 1-11 (2004).
- L. Snyder, J. Dolan, and J. Gant, J. Chromatogr. A.165, 3-30 (1979).
- J. Stranahan and S. Deming, Anal. Chem.54, 1540-1546 (1982).
- J. Dolan, LCGC 6, 112-116 (1988).
- J. Ermer, J. Pharm. Biomed. Anal.18, 707-714 (1998)
- J. Dolan, J. Kern, and T. Culley, LCGC 14, 202-208 (1996).
- M. Nelson and J. Dolan J, LCGC 16(11), 992-996 (1998).
- J. Dolan, LCGC N. Amer. 31(8), 604-611 (2013).
- S. Mabic, C. Regnault, and J. Krol, LCGC North Amer.1, 1-5 (2005).
- M. Garcia, J. Chromatogr. B.825, 111-123 (2005).
- S. Liu, Am. Lab.40, 22-29 (2008).
- P. Sadek, The HPLC Solvent Guide (2nd Edition, John Wiley and Sons, Inc, New York, USA, 2001).
- A. Kaufmann and P. Butcher, Rapid. Commun. Mass Spectrom.19, 3694-3700 (2005).
- W. Siegert, A. Guckel, and S. Carstens, Int. J. App. Sci.137, 35-40 (2011).
- R. Rudel and L. Perovich, Atmos. Environ.43(1), 170-181 (2009).
- W. Li and J. Duan, RSC: Analytical Methods3, 314-321 (2010).
- A. Reid, C. Brougham, A. Fogarty, and J. Roche, Int. J. Environ. Anal. Chem.87, 125-133 (2007).
- V. Lopez-Avila, J. Benedicto, J. Milanes, and W. Beckert, J. Assoc. Off. Anal. Chem.73, 709-720 (1990).
- M. Ende and G. Spiteller, Mass Spectrom. Rev.1, 29-62 (1982).
- C. Huck C and G. Bonn, J. Chromatogr. A885, 51-72 (2000).
- M. Shaw, G. Eaglesham, and J. Mueller, Chemosphere75, 1-7 (2009).
- K. Ensing, J. Franke, A. Temmink, X. Chen, and R. De Zeeuw, J. Forensic Sci.37, 460-466 (1992).
- M. Ringo, J. Allen, and D. Mattocks, LCGC North Am.21, 167-170 (2003).
- S. Kulkarni, J. George, and V. Joshi, Am. Biotechnol. Lab.26, 1-2 (2008).
- I. Henderson, R. Norhaus, and J. Anderson, J. Chromatogr. A. 546, 61-71 (1991).
- P.L. Zhu, L.R. Snyder, and J.W. Dolan, J. Chromatogr. A 718, 429 (1995).
- L. Du, D. Musson, and A. Wang, Rapid Commun. Mass Spectrom.19, 1779-1787 (2005).
Mark Taylor has worked in analytical method development for 30 years and is currently lab manager for the Project Analytics group in Analytical R+D at Pfizer, Sandwich, UK. Prior to joining Pfizer Mark worked at the Horseracing Forensic Laboratory in Newmarket, UK, for 13 years. Mark studied for his PhD at St Bartholomew’s College, London, UK, under the supervision of Professor David Perrett specializing in forensic applications of capillary separation techniques. Mark is a visiting lecturer at Kings College London, UK, and has a keen interest in promoting best practices in chromatography to anyone who will listen.
Fiona Harvey-Doyle has a degree in applied chemistry and graduated from Loughborough University with an MSc and Ph.D in analytical chemistry under the supervision of Prof. Roger Smith. She then moved to Sandwich where she has been working for Pfizer for the past 7 years. Fiona has worked in several roles for Pfizer as a project lead in the early phase portfolio and more recently as a drug product lead in late stage development supporting late stage filings and commercial products.
Krina Patel completed her undergraduate degree in pharmaceutical sciences at Kingston University in 2011 and then went on to study at King’s College London for a master’s degree in analytical science for industry. Her MSc degree involved a project placement with the analytical research and development team at Pfizer in Sandwich, UK. Krina is now in her final year of her PhD at the University of Southampton focusing on the analysis of deposit formation in diesel engines using a range of analytical techniques including GC-MS, LC-MS, and SFC-MS, with an expected completion date of January 2016.





; An Interview with Fabrice Gritti")
: An Interview With Fabrice Gritti")

. | Image Credit: © Joan Vadell - stock.adobe.com")
Detecting Hyper-Fast Chromatographic Peaks Using Ion Mobility Spectrometry
May 6th 2025Ion mobility spectrometers can detect trace compounds quickly, though they can face various issues with detecting certain peaks. University of Hannover scientists created a new system for resolving hyper-fast gas chromatography (GC) peaks.


University of Oklahoma and UC Davis Researchers Probe Lipidomic Profiles with RP-LC–HRMS/MS
May 6th 2025A joint study between the University of Oklahoma Health Sciences Center (Oklahoma City, Oklahoma) and the UC Davis West Coast Metabolomics Center (Davis, California) identified differentially regulated lipids in type 2 diabetes (T2D) and obesity through the application of reversed-phase liquid chromatography-accurate mass tandem mass spectrometry (RP-LC-accurate MS/MS).


Automated Sample Preparation (ISO 20122) for MOSH/MOAH in Seasoning Oils
May 6th 2025This work presents an Automated Sample Preparation procedure for MOSH/MOAH analysis of Seasoning Oils. We compare results from a manual epoxidation procedure compliant with DIN 16995 with results based on fully automated sample preparation (epoxidation and saponification) compliant with ISO 20122. In both cases, online clean-up via activated aluminum oxide (AlOx) are used to remove interfering n-alkanes from the MOSH fraction during the HPLC run. Automated data evaluation using a dedicated software (GERSTEL ChroMOH) is presented.

