Pressure, Particles, and System Contribution: The Holy Trinity of Ultrahigh-Performance Liquid Chromatography
LCGC Europe
The last decade has witnessed how liquid chromatography columns and instruments changed from long bulky columns with relatively large fully porous particles operated at modest pressures (100–200 bar), to short compact columns with small superficially porous particles operated at ultrahigh pressures (1200–1500 bar). This (r)evolution has resulted in a tremendous increase in achievable separation performance or decrease in analysis time, but requires a good knowledge of optimal chromatographic conditions for each separation problem and, concomitant, the right instrument configuration.



Ken Broeckhoven, Jelle De Vos, and Gert Desmet, Vrije Universiteit Brussel, Department of Chemical Engineering, Brussels, Belgium
The last decade has witnessed how liquid chromatography columns and instruments changed from long bulky columns with relatively large fully porous particles operated at modest pressures (100–200 bar), to short compact columns with small superficially porous particles operated at ultrahigh pressures (1200–1500 bar). This (r)evolution has resulted in a tremendous increase in achievable separation performance or decrease in analysis time, but requires a good knowledge of optimal chromatographic conditions for each separation problem and, concomitant, the right instrument configuration.
The improvements in separation power achieved over the past decade (a 10-fold decrease in separation time and threefold increase in efficiency) were only possible through the combined advancement of three aspects of chromatographic technology: improved columns with smaller particles, higher operating pressures, and systems with decreased extracolumn dispersion (1). From a column technology perspective, the introduction of smaller (sub-2-µm) particles made it possible to achieve the same separation efficiency in a much shorter timescale, proportional to the square of the inverse particle size (1/dp²) (2). In addition, the introduction of so-called superficially porous particle (SPP) (also known as core–shell or solid core) columns that achieve reduced plate heights 25% lower than traditional fully porous particles (FPP) made a reduction in analysis time of 40% or an increase in efficiency of 25% possible (3). To operate smaller particles in reasonable column lengths (L) at their optimal velocity (uopt), the introduction of equipment able to operate at pressures beyond the traditional 400 bar limit of high performance liquid chromatography (HPLC) instrumentation (ultrahigh-performance liquid chromatography [UHPLC] pressures were originally up to 1000 bar and are now up to 1500 bar) and sufficiently wellâpacked columns to withstand the steeper and higher pressure cycles were required. Typically, shorter columns (3–10 cm) are used versus the traditional HPLC columns (5–25 cm) because of the higher efficiencies obtained with smaller particles. To manage the viscous heating effects that prevail at elevated pressure drops (4), in addition to reducing solvent consumption at the higher optimal flow rates for smaller particles, the internal diameter (i.d.) of the columns was also reduced from 4.6 mm for HPLC to 2.1 mm for UHPLC columns. This reduction in column volume requires a concomitant smaller instrument contribution to band broadening to maintain the separation quality. Therefore, in order to achieve ultrahigh-performance, it is required to have both state-of-the art instrumentation with a high operating pressure and novel columns (2,5). In this article, the advances made over the last decade in (U)HPLC are illustrated, the limitations from maximum instrument pressure and instrument contribution highlighted, and the possibilities for further development in instrumentation and operating pressure investigated.
Performance Improvements as a Result of Particle Size and Morphology

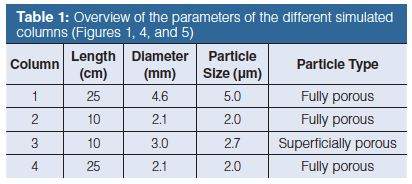

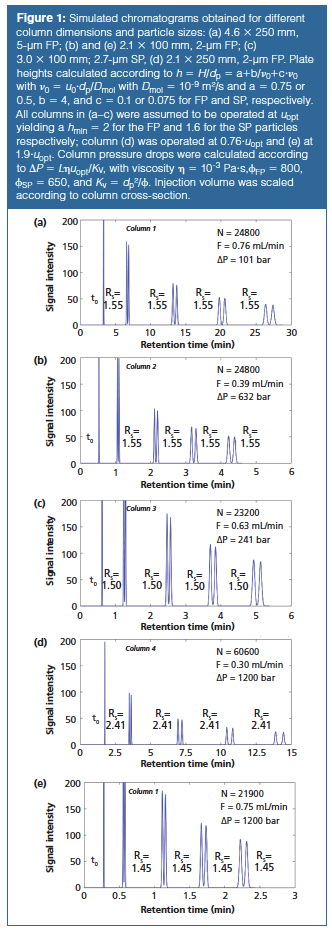
Figure 1(a–c) shows simulated chromatograms of the separation of 4 peak pairs (first component of each at k = 1, 3, 5, and 7 and selectivities of 1.080, 1.053, 1.048, and 1.046, respectively to obtain the same resolution Rs). Column 1 (Figure 1[a]) has the typical dimensions of routinely used columns on HPLC instrumentation, that is, 25-cm long with a 4.6 mm i.d. and packed with 5-µm FPPs (see Table 1 for an overview of the properties of the different columns). Column 2 (Figure 1[b]) represents its UHPLC equivalent (same plate count) of 10-cm long, but packed with 2-µm FPPs, each operated at its optimum velocity and assuming a reduced plate height h = H/dp = 2. It becomes immediately clear that the same separation efficiency and resolution is achieved in both separations, but 6.25 times faster on Column 2. The latter separation, however, also requires the same increase in operating pressure, exceeding that of standard HPLC instrumentation (400 bar). The corresponding flow rate for Column 2 was calculated assuming a narrow internal diameter (2.1 mm) column typically used in UHPLC. As mentioned earlier, besides a reduction in solvent consumption, these narrower internal diameter columns are required to compensate for the increased viscous heating effects that occur at elevated pressure drops required to operate them at or above their optimal velocity. A good compromise between gain in analysis time and increase in pressure drop can be obtained when switching from a FPP column to a column packed with 2.7-µm SPPs (Figure 1[c], Column 3). These particles not only typically have minimum reduced plate heights closer to 1.5, but also, because of the lower porosity and lower flow resistance Ï (15–20%) require lower pressure drops for similar velocities. As a result, with much larger particles (2.7 µm versus 2 µm) roughly the same efficiency (-6.5%) can be obtained for only a small increase in analysis time (+17%), as can be seen when comparing Figure 1(c) (Column 3) with Figure 1(b) (Column 2). The main advantage lies in the much lower operating pressure for the 2.7-µm particles (versus the 2 µm particles) required to run this column at its optimum flow rate (3). The required pressure is in fact only 2.4 times higher than for the 5-µm FP particles. For this reason a 3 mm i.d. was considered for Column 3 because the issue of viscous heating at these lower pressures is much less pronounced (3). Recently, it was demonstrated that FPPs with a narrow particle size distribution can reach minimum reduced plate heights of 1.7–1.9, that is, in between those of traditional fully porous (h = 2) and SPPs (h = 1.5) (6).
Gains in Analysis Time and Efficiency With Operating Pressure
Figure 1 shows how the same separation quality is achieved in a shorter time by changing particle type and size. However, it is also possible to keep the same analysis time (or have a smaller gain in analysis time), but increase the separation quality by using longer columns. Figure 1(d) shows the separation on a 25-cm long column with 2-µm FPPs (Column 4). If run at the optimal velocity, this would require an operating pressure above 1500 bar. To represent more realistic conditions, the flow rate was limited to that corresponding with a pressure limit of 1200 bar (which all UHPLC instruments can currently reach), resulting in a velocity slightly (-24%) below the optimal velocity. Nevertheless, the 2-µm particles can be operated at twice the velocity as the 5-µm particles (2.5 times faster in the absence of the pressure limitation), yielding a reduction in analysis time of a factor of two, a gain in efficiency of 2.4, and an increase in resolution of 1.6 (since RS ~ N0.5). An additional benefit of using small particles is that they can be operated at velocities above their optimum and only experience a small loss in performance because the C-term contribution to H is proportional to dp2. This is illustrated in Figure 1(e), where Column 2 is operated at almost twice (1.9×) its optimal velocity (limited to ΔP = 1200 bar). Whereas little or no efficiency and resolution is lost, the separation is performed almost twice as fact.

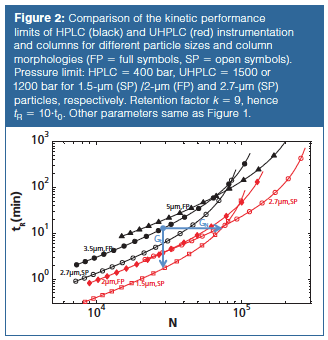
To combine the optimization of mobile phase velocity, column length, and particle size and at the same time take into account the pressure limitations of a system or column, the so-called kinetic plot methodology (7–11) provides a more concise way to compare the separation performance in (U)HPLC. By plotting the kinetic performance limits (KPL), that is, the shortest possible time to reach a certain performance, or, equivalently, the highest possible performance that can be reached for a given analysis time, the most optimal conditions for each chromatographic support (particle size and type) are compared (8,10). These limits are obtained by operating at the maximum pressure (400 bar for HPLC, 1200–1500 bar for UHPLC) and optimizing the column length. Such KPLs are plotted in Figure 2(a), which is an updated version (simplified) of figure 1 in reference 9 and figure 3 in reference 12. The figure provides an historic overview of the progress made from HPLC (black, 3.5–5-µm FPP, and 2.7-µm SPP) towards UHPLC particles and conditions (red, 2-µm FPP, 1.5- and 2.7-µm SPP) by plotting analysis time tR (calculated as tR = t0•(1 + k) with k = 9) versus efficiency N. The advantage of a higher operating pressure is clear because the red curves are located lower (faster analysis) and more to the right (higher efficiency) (8,9). In addition, the higher operating pressure favours the use of smaller particles. For HPLC, the advantage of SPPs clearly stands out because they outperform 3.5-µm particles over the entire relevant range of efficiencies (the range where 3.5-µm particles have a better kinetic efficiency than 5-µm particles) (8). A similar conclusion can be drawn for the UHPLC separations, where the SPPs outperform their FPP counterparts, even though for the 2.7-µm particles a lower pressure limit (1200 bar) is assumed than for the 2-µm FPPs (where we took 1500 bar-currently the highest commercially available instrument pressure-as for the 1.5 µm SPPs). It can also be shown that the benefits of an increase in operating pressure and the use of more efficient superficially porous columns are additive. This methodology also allows kinetic gain factors that quantify the gain in analysis time (Gt) (for the same N) or efficiency (GN) (for the same t) that can be reached by switching from one (old) operating pressure, particle type, and size to another (new) combination to be defined (1,9).

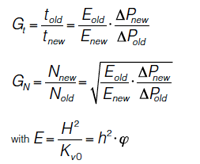
[1]
In these expressions, E represents the separation impedance determined by the square of the plate height H (at the chosen flow rate) divided by the column permeability Kv0 based on the velocity u0 of an unretained compound (Kv0 = dp²/Ï, with Ï the flow resistance). For the arrows given in Figure 2 (Nold = 30000, tR,old = 13.6 min), both starting (ΔPold = 400 bar) and ending (ΔPnew = 1500 bar) on a kinetic plot, the gains when switching from HPLC with 3.5-µm FPPs to UHPLC with 1.5âµm SPPs are Gt = 7.6 (tR,new = 1.8 min) and GN = 2.48 (Nnew = 74500). For these analysis times and conditions, the 3.5âµm FPP column at 400 bar for tR = 30 min is operated at a velocity close to the optimum regime (h = 2.12; E = 3596). This is also the case for the 1.5 µm particles operated at 1500 bar such that tR = 2.79 min (h = 1.66; E = 1791). For the 1.5 µm tR = 13.6 min case, slightly larger particles would be better suited because the column is operated just below the optimal velocity, that is, in the B-term regime (h = 1.84; E = 2200).

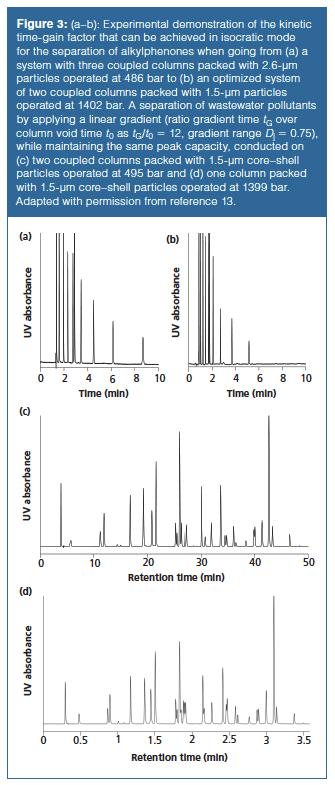
Figure 3(a) and 3(b) represent results from an experimental investigation of these gain factors in isocratic mode (13). By switching from three coupled 10-cm columns (30 cm) packed with 2.6-µm core–shell particles operated at almost 500 bar (Hold = 5.46 µm, Kv0 = 14.8 10-15 m2, Figure 3[a]) to two coupled 10-cm columns (20 cm) with 1.5-µm core–shell particles operated at 1402 bar (Hnew = 3.96 µm, Kv0 = 3.8 10-15 m2, Figure 3[b]), a nearly equivalent separation efficiency was measured (Nold = 54,900 versus Nnew = 50,500, respectively), but a kinetic time gain factor of 1.6 was achieved. The gain in analysis time by a factor of 1.6 is slightly higher than the theoretically calculated value, which is only Gt = 1.4, but this theoretical value assumes that exactly the same efficiency N is reached for both separations. However, as the separation efficiency in the improved (“new”) system is around 10% lower than for the “old” system because of practical limitations (available column lengths), a larger gain in analysis time is found for slightly less efficiency. To obtain the same N, a slightly longer column (~10% larger, corresponding to a 22-cm column) should have been used at a slightly lower flow rate (also ~10% smaller), resulting in a time gain closer to 1.4 than the experimental value of 1.6. Figure 3(c) and 3(d) demonstrate the kinetic-time gain that can be achieved by optimizing a gradient separation of wastewater pollutants. A linear gradient from 20:80% (v/v) acetonitrile–water to 95:5% (v/v) acetonitrile–water was applied with tG/t0 = 12. By using 1.5âµm core–shell particles, and going from two coupled columns operated at 500 bar (Figure 3[c]) to one column operated at 1400 bar (Figure 3[d]), a kinetic timeâgain factor of almost 13 was achieved. Although almost exactly the same peak capacity of around 190 was achieved (13), some differences in selectivity and resolution can be seen on the two chromatograms. These deviations are the result of pressure and temperature (viscous heating effects) gradients that induce changes in retention factor that can affect separation quality (4,14,15).
Requirements Regarding Instrumentation Contribution to Dispersion

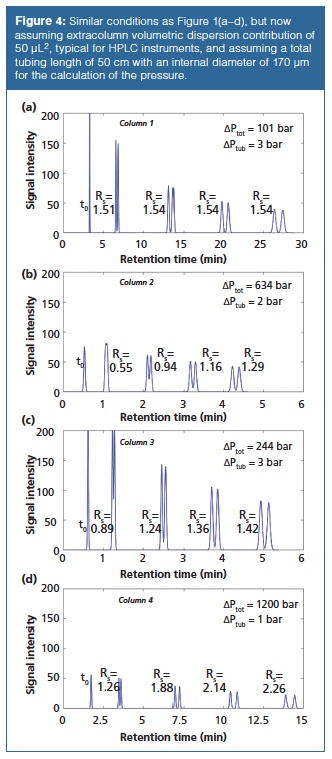
For the sake of comparison, it was assumed that for the separations in Figure 1 the instrument did not contribute to the overall band broadening. However, it is obvious that the size of the injection plug (volume), diameter of the connection tubing, and volume of the preheater and detection cell all have an impact on the separation efficiency (16). Figure 4 therefore represents the same column performance (N ⊕ 25000) as considered in Figure 1, but assuming an additional system contribution to dispersion of 50 µL2, which is a typical value for a standard HPLC instrument (5,17) and assuming connection tubing with an internal diameter of 170 µm and a total length of 50 cm to calculate the total system operating pressure. Whereas for Column 1 (the long wide-bore column packed with large 5-µm particles) little or no effect of extracolumn dispersion can be observed, the separation quality on the shorter narrow bore Column 2 with 2-µm particles is completely gone, showing a complete overlap for the early eluting pair and still a significant loss in resolution for late-eluting compounds. Since both columns are operated on the same system, Column 2 is much more affected because it has a much smaller volumetric dispersion as a result of the smaller column volume (1/12th) (18). For Column 3, packed with SPPs, the loss in performance from extracolumn dispersion is still significant, but smaller because a larger internal diameter (3 mm) can be used. The large internal diameter of the connection tubing ensures that little or no additional pressure is required at the represented flow rates. For the case of Column 4 (25-cm long with 2-µm particles), a significant loss in resolution is observed, but, as the longer and thus larger volume column is less affected by extracolumn dispersion and the superior performance already resulted in a higher resolution than necessary, the baseline separation (RS = 1.5) is only compromised for the first eluting pair. Figure 4 illustrates how the switch to smaller particles or more efficient particle morphologies (SPPs) alone is not sufficient to obtain a better separation performance, but that the dispersion in the chromatographic system also needs to be considered (5).

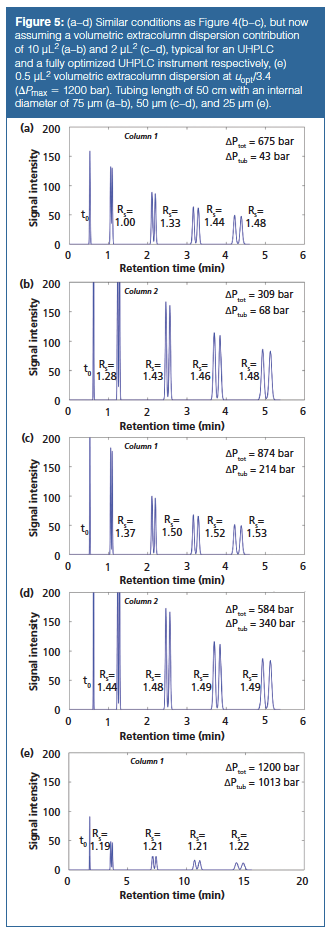
Figures 5(a) and 5(b) show the obtained performance for Column 2 and Column 3 (the performance of Column 1 was only negligibly affected by the system and is not shown), but now with the typical dispersion (17) for a UHPLC instrument-10 µL2-and assuming this requires the same tubing length as in Figure 4 (50 cm) but now with an internal diameter of 75 µm. A significant improvement of the separation, especially for the early-eluting compounds, can be observed (18–21). Nevertheless, the early-eluting compounds are not yet baseline resolved. In addition, a significant increase in operating pressure from the narrower tubing is obtained. This increase is larger for Column 3 packed with SPPs because of the larger flow rate (see Figure 1). To get the full benefits from the narrow internal diameter columns, it is thus required to operate them on a fully optimized UHPLC system with reduced extracolumn dispersion. A number of groups have reduced the extracolumn dispersion to values as low as 2 µL2 (20,21,22,23). This allowed them to obtain more than 90% of the intrinsic column efficiency for retention factors > 3. This is illustrated in Figures 5(c) and 5(d), which show little or no loss in separation resolution for all peaks except at k = 1. However, there is a steep increase in operating pressure as a result of the reduced internal diameter of the connection tubing. Once again, this is more pronounced for the 3-mm i.d. column (Column 3) where more than 60% of the total pressure drop is a result of the connecting tubing (versus around 30% for Column 2, with a 2.1 mm i.d.). In fact, for this case, the further decrease in extracolumn dispersion results in only a small gain in resolution but at a large cost in pressure drop. When a reduction in extracolumn volume results in such a cost in pressure drop that column length or flow rate has to be decreased, it will never be advantageous from a performance perspective. As an example, Figure 5(e) assumes connecting tubing of 25 µm i.d. and a system dispersion of 0.5 µL2 for Column 2. The excessive pressure drop (at a fixed flow rate ΔPtub~dtub4) meant that the mobile phase velocity had to be decreased by a factor of 3.4 and therefore the column was operated far into the B-term regime. As can clearly be seen, this results in a significant increase in analysis time and a decrease in separation resolution. As an alternative (results not shown), one could opt for a 4.6 mm i.d. column packed with 2.7-µm SPPs, in which case even on the HPLC system with 50 µL2 a resolution of 1.29 is found for the first peak pair, but, with the 170-µm tubing, only an operating pressure of 247 bar is required. The price to be paid is however a higher solvent consumption because the flow rate increases by a factor of around 2.4 and, although the pressure drop is limited, viscous heating related performance can become more pronounced if larger internal diameter columns are used.
Examples of System Contributions in Gradient and Isocratic Elution

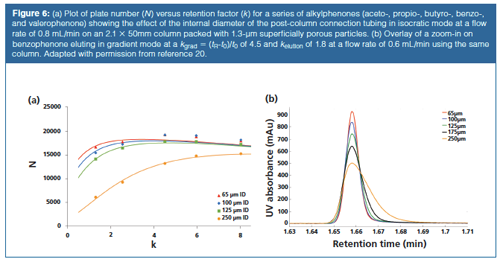
Figure 6(a) and 6(b) illustrate how the optimization of extracolumn volumes can affect separation performance in both (a) isocratic and (b) gradient separations (20). In this case, a small volume column (2.1 × 50 mm) packed with very small SPPs (1.3 µm) was run on a fully optimized chromatographic system (on-column focusing with POISE [24], 80 nL UV detector cell, minimized pre-column tubing) except for the short (14 cm) tubing connecting the column end with the detector cell. For this tubing, different internal diameters were chosen from 65 to 250 µm. For the isocratic case, the variation of the obtained separation efficiency (represented by the plate number N) versus retention factor is plotted. Even if only one part of the flow path-in this case the short 14-cm tubing from the column to the detector-is not optimized, a significant loss in performance already occurrs as for k < 4 a much lower efficiency than the expected N = 20,000 is found. As was illustrated in the previous simulated chromatograms, the first eluting compounds suffer most from the extracolumn band broadening (18,19,21). The slight downward trend in efficiency at higher retention factors results from the effect of retention factors on plate height and optimum velocity (20). In Figure 6(b), a zoom-in on one peak (benzophenone) in gradient elution mode is shown for the different tubing internal diameters. This compound elutes around an (apparent) gradient retention factor kgrad of 4.5. It can readily be seen that the wider tubing has a significant impact on the peak widths and heights. It is also clear that, contrary to what is often assumed, extracolumn band broadening can also have a significant impact in gradient mode because post-column dispersion is not minimized as a result of the typical on-column focusing at the start of a gradient run. For example, Spaggiari et al. demonstrated that when coupling UHPLC with MS, the interface tubing has to be minimized to have negligible impact on performance for k values greater than 7 for a standard system (25) and optimal MS settings need to be applied (18,25).
Conclusions
Faster or better separations can be obtained by switching to smaller particles and by using superficially porous particles. However, smaller particles require higher operating pressures. To avoid pressure drop limitations caused by viscous heating and to reduce solvent consumption, these small particles are packed in short columns with a narrower internal diameter. As a consequence, the performance of these small volume columns is strongly affected by the extracolumn dispersion in the chromatographic system. This article has shown that all three cornerstones of UHPLC need to be improved to achieve the expected higher separation performance: smaller and better particles, higher operating pressures, and reduced system contributions.
References
- J. De Vos, K. Broeckhoven, and S. Eeltink, Anal. Chem.88, 262–278 (2016).
- S. Fekete, E. Oláh, and J. Fekete, J. Chromatogr. A 1228, 57–71 (2012).
- J. Ruta, D. Zurlino, C. Grivel, S. Heinisch, J.-L. Veuthey, and D. Guillarme, J. Chromatogr. A1228, 221–231 (2012).
- D. V. McCalley, TrAC Trends Anal. Chem.63, 31–43 (2014).
- K.J. Fountain, U.D. Neue, E.S. Grumbach, and D.M. Diehl, J. Chromatogr. A1216, 5979–5988 (2009).
- O.H. Ismail, M. Catani, L. Pasti, A. Cavazzini, A. Ciogli, C. Villani, D. Kotoni, F. Gasparrini, and D.S. Bell, J. Chromatogr. A1454, 86–92 (2016).
- K. Broeckhoven, D. Cabooter, and G. Desmet, LCGC Europe24, 396–404 (2011).
- K. Broeckhoven, D. Cabooter, S. Eeltink, and G. Desmet, J. Chromatogr. A1228, 20–30 (2012).
- K. Broeckhoven and G. Desmet, TrAC Trends Anal. Chem.63, 65–75 (2014).
- P.W. Carr, X. Wang, and D.R. Stoll, Anal. Chem.81, 5342–5353 (2009).
- G. Desmet, D. Cabooter, and K. Broeckhoven, Anal. Chem.87, 8593–8602 (2015).
- R.E. Majors, LCGC Europe28, 658–665 (2015).
- J. De Vos, M. De Pra, G. Desmet, R. Swart, T. Edge, F. Steiner, and S. Eeltink, J. Chromatogr. A1409, 138–145 (2015).
- G. Vanhoenacker, F. David, B. Blatz, E. Naegele, and P. Sandra, “Increasing Productivity in the Analysis of Impurities in Metoclopramide Hydro-chloride Formulations Using the Agilent 1290 Infinity LC System”, Application note, (2009) http://cp.chem.agilent.com/Library/applications/5990-3981EN.pdf.
- K. Broeckhoven, J. Billen, M. Verstraeten, K. Choikhet, M. Dittmann, G. Rozing, and G. Desmet, J. Chromatogr. A1217, 2022–2031 (2010).
- J.P. Grinias, B. Bunner, M. Gilar, and J.W. Jorgenson, Chromatography2, 669–690 (2015).
- S. Fekete and J. Fekete, J. Chromatogr. A1218, 5286–5291 (2011).
- S. Buckenmaier, C.A. Miller, T. van de Goor, and M.M. Dittmann, J. Chromatogr. A1377, 64–74 (2015).
- N. Lambert, S. Miyazaki, M. Ohira, N. Tanaka, and A. Felinger, J. Chromatogr. A1473, 99–108 (2016).
- K. Vanderlinden, K. Broeckhoven, Y. Vanderheyden, and G. Desmet, J. Chromatogr. A, 1442, 73–82 (2016).
- F. Gritti, T. McDonald, and M. Gilar, J. Chromatgr. A1420, 54–65 (2015).
- K.J. Fountain and P.C. Iraneta, in UHPLC in Life Sciences, No. 16, D. Guillarme and J.-L. Veuthey, Eds., (RSC Chromatography monographs, Cambridge, UK, 2012), pp. 29–66.
- F. Gritti, T. McDonald, and M. Gilar, J. Chromatogr. A 1420, 54–65 (2015).
- A.C. Sanchez, J.A. Anspach, and T. Farkas, J. Chromatogr. A1228, 338–348 (2012).
- D. Spaggiari, S. Fekete, P.J. Eugster, J.L. Veuthey, L. Geiser, S. Rudaz, and D. Guillarme, J. Chromatogr. A1310, 45–55 (2013).
Ken Broeckhoven is an associate professor in the Department of Chemical Engineering at the Vrije Universiteit Brussel, Brussels, Belgium. His research focuses on the development of novel ultrahighâpressure liquid chromatography instrumentation, fundamental aspects of supercritical fluid chromatography (SFC), investigation of the parameters affecting column performance, and the modelling of flow effects in chromatographic systems.
Jelle De Vos is a postdoctoral researcher at the Department of Chemical Engineering at the Vrije Universiteit Brussel. He is currently developing novel multilayered rotor technology to advance multidimensional separations and establishing microfluidic chip technology for threeâdimensional spatial liquid chromatography.
Gert Desmet is a full professor in biochemical and chemical engineering at the Vrije Universiteit Brussel. His research focuses on the miniaturization of separation methods and on the investigation of flow and mass transfer effects in chromatographic systems. He is the first or senior author of over 250 peer-reviewed papers and 10 patent applications.

.jpg")

.jpg")
.jpg")
.jpg")
.jpg")
.png")
.png")
.png")
.png")

.png")





New Study Reviews Chromatography Methods for Flavonoid Analysis
April 21st 2025Flavonoids are widely used metabolites that carry out various functions in different industries, such as food and cosmetics. Detecting, separating, and quantifying them in fruit species can be a complicated process.


Extracting Estrogenic Hormones Using Rotating Disk and Modified Clays
April 14th 2025University of Caldas and University of Chile researchers extracted estrogenic hormones from wastewater samples using rotating disk sorption extraction. After extraction, the concentrated analytes were measured using liquid chromatography coupled with photodiode array detection (HPLC-PDA).



