Myths in Ultrahigh-Pressure Liquid Chromatography
The advent of ultrahigh-pressure liquid chromatography (UHPLC) and its successful commercialization in the last few years has brought forth a modern high performance liquid chromatography (HPLC) platform capable of higher speed, resolution, precision, and sensitivity. Currently, all major HPLC manufacturers have some type of low-dispersion UHPLC products with upper pressure limits ranging from 15,000 to 19,000 psi (1000 to 1300 bar) on the market. This installment describes a number of popular myths or half-truths in UHPLC and provides data that contradict or even repudiate some of these commonly held beliefs.
The advent of ultrahigh-pressure liquid chromatography (UHPLC) and its successful commercialization in the last few years has brought forth a modern high performance liquid chromatography (HPLC) platform capable of higher speed, resolution, precision, and sensitivity. Currently, all major HPLC manufacturers offer some type of low-dispersion UHPLC products with upper pressure limits ranging from 15,000 to 19,000 psi (1000 to 1300 bar). This instalment describes a number of popular myths or half-truths in UHPLC and provides data that contradict or even repudiate some of these commonly held beliefs.
For five decades since the 1960s, the pressure limits of high performance liquid chromatography (HPLC) systems remained stagnant at 6000 psi (400 bar). This pressure limit was appropriate for the column packings available at the time, which continuously trended towards smaller particle sizes (that is, from 30, 10, 5, to 3 µm). There appeared to be no concerted efforts to increase the system pressure ratings during that period with the exception of an exploratory study published in 1969 (1). The "breakthrough" in ultrahigh-pressure liquid chromatography (UHPLC) came in 1997 with proof-of-concept research by James Jorgenson (2) and follow-on studies by Milton Lee (3). These early studies demonstrated spectacular performance (column efficiency, N = 200,000 plates) at very high pressures (>60,000 psi) in research systems using capillary columns. However, the impact of their discoveries for typical practitioners and for routine applications were only possible after the debut of commercial UHPLC equipment with reliable autosamplers and gradient capabilities
In 2004, Waters Corporation introduced the first UHPLC system — the Acquity UPLC (Ultra-Performance LC) system with an upper pressure limit of 15,000 psi (1000 bar) together with Acquity UPLC columns (1.0 and 2.1 mm i.d.) packed with sub-2-µm hybrid particles (4–8). Although this pressure rating was modest in comparison to that achieved using the early research systems, the new UHPLC system generated considerable excitement and established higher performance benchmarks and expectations. These early systems enjoyed immediate acceptance in research applications despite some initial concerns over injection precision and other issues in quality control (QC) applications (7,9). Other manufacturers quickly followed with their own UHPLC systems. By 2010, the transformation from HPLC to UHPLC was essentially complete with UHPLC product offerings available from most major vendors. Today, all UHPLC systems have reduced system dispersion and dwell volumes as well as improved precision and sensitivity (10).
The fundamentals, benefits, potential issues, and best practices of UHPLC are well documented (5–7, 9–15). Some of the key benefits are as follows:
- Faster analysis with good (or acceptable) resolution — the primary incentive for new users in high-throughput screening, liquid chromatography–mass spectrometry (LC–MS), routine testing, and method development.
- Superiority in high-resolution separations of complex samples — peak capacities of 600–1000 are now possible in a reasonable time (<60 min under gradient conditions). This capability is transformative in life science research and the analysis of complex pharmaceuticals, filling an unmet need for QC applications (13–15).
- Other benefits of UHPLC versus conventional HPLC include substantial solvent savings (5- to 15-fold), increased mass sensitivity in UV detection (3–10-fold), and improved precision for both retention times (2- to 3-fold) and peak areas (<0.1% RSD).
In the last few years, UHPLC has evolved from a scientific curiosity for early adopters in research and high-throughput screening into a modern standard HPLC platform. As the saga of UHPLC unfolded, a number of myths or half-truths have emerged. The goal of this column instalment is to describe some of the more interesting myths and provide evidence to delineate or repudiate these widely held misconceptions. The myths:
- You don't need an expensive UHPLC system — high-temperature LC or core–shell columns will get you there.
- Viscous heating is a "huge" issue for sub-2-µm particle columns.
- A 2.1-mm i.d., sub-2-µm column is the best choice for UHPLC.
- Gold-plated fittings with double ferrules are needed in UHPLC.
- A binary high-pressure mixing pump is a "must".
- UHPLC provides substantially higher UV sensitivity than conventional HPLC.
- Method transfer between UHPLC and HPLC is very easy ("a piece of cake") and method revalidation is not needed.
- Lower-dispersion UHPLC systems are better.
Dispelling Some Popular Myths in UHPLC
You don't need UHPLC — high-temperature LC or core–shell columns will get you there: In April of 2010, I was invited to a local meeting to give a presentation on UHPLC. The format turned out to be a debate between two opposing viewpoints on UHPLC versus high-temperature LC. I remembered being surprised by some comments that UHPLC was a marketing hype invented by the vendors to extract more money from the user. At first, I thought that this conspiracy theory was a joke but it turned out to be serious. I also recalled hearing this line of argument about high-temperature LC around 2006, mostly from vendors in the "have not" camps that indicated high column temperatures will allow one to use small-particle columns without a major capital investment. Along the same line are comments that superficially porous sub-3-µm (core–shell) material is a more cost-effective alternative to expensive UHPLC systems.
Nowadays, most practitioners may realize that these arguments are not valid because UHPLC can be used in combination with these approaches (including two-dimensional [2D] LC) with superior results, as they are options rather than alternatives (13,14). The use of high-temperature LC above 60–70 °C is not viable for thermally labile pharmaceuticals and compounds (16,17). Core–shell (also known as fused core, solid core, or superficially porous) material is becoming the dominant contender to totally porous material for all applications (18). However, the notion that core–shell columns will lessen the need for UHPLC is less compelling with recent introductions of 1.3- and 1.6-µm core–shell particles that deliver ~400,000 plates/m (18). I believe the availability of sub-2-µm core–shell material represents an exciting advancement in column technology. With overwhelming increases in efficiency over fully porous material demonstrated in initial studies (+40% versus 1.7-µm particles and >200% versus 3.0-µm fully porous particles), its impact can be transformative in modern HPLC.
Today, the objections to UHPLC versus high-temperature LC or core-shell columns by skeptics are waning as UHPLC is becoming a mainstream platform.
Viscous heating is a "huge" issue for sub-2-µm particle columns: A frictional heating phenomenon is observed when the mobile phase is pumped at a relatively high flow rate and operating pressure through columns packed with very small particles. The heat generated is cumulative, giving rise to longitudinal thermal gradients along the length of the column. The heat is simultaneously dissipated through the column wall, resulting in radial thermal gradients and parabolic flow profiles that cause band broadening. This is a popular research topic with dozens of papers already published (19–21). These complex effects are dependent on the type of column oven, particle size, column length and diameter, thermal conductivity of the mobile phase, and flow rate.
It turns out that radial thermal gradients are indeed problematic when the column wall temperature is controlled under isothermal conditions (that is, in a water bath or to some extent in a forced air column oven). For still-air column ovens, the longitudinal heating effect is more serious and can increase the temperatures at the end of the column by 10 °C to 20 °C (19,20). Although this does not cause band broadening, it raises the average temperature of the column, causing lower retention and potential selectivity changes. In situations where HPLC method conditions are transferred to UHPLC, this effect can be partially mitigated by deliberately setting the UHPLC methods to a lower column temperature (for example, 5 °C). Another viable solution to reduce the effect of longitudinal heating is to introduce intermediate active cooling by connecting shorter columns together to form a longer column (22). This longitudinal heating effect may cause issues when transferring methods, particularly across UHPLC platforms from different vendors and with varying types of column ovens.
For most users, it is important to acknowledge the existence of viscous heating, however, it may not be a serious practical issue except for columns packed with very small particles (<1.5 µm), operation at very high pressures (>800 bar), or with a forced air oven and if there are critical pairs sensitive to temperature changes.
A 2.1-mm i.d., sub-2-µm column is the best choice for UHPLC: The most popular UHPLC column format consists of 2.1-mm i.d. columns packed with sub-2-µm particles; these columns are particularly well-matched to the first commercial systems in 2004 (6). Since then, there has been a trend towards UHPLC systems to accommodate existing HPLC methods with larger-diameter columns and a higher flow rate range (>2 mL/min), a bigger column oven (>150 mm), larger injection loops (>20 µL), and larger internal diameter connection tubing. These UHPLC systems would have higher system dispersion, a trade-off for better flexibility and compatibility to existing HPLC methods.
Figure 1 compares the isocratic performance of three 50-mm long columns of various diameters (2.1, 3.0, and 4.6 mm) packed with 1.8-µm C18 particles with appropriate flow adjustments. Note that the 4.6-mm i.d column displays significantly higher column efficiencies (N = 12,860 versus 8170 for the 2.1-mm i.d. column). This observation is in line with the notion that the detrimental "wall effect" is more pronounced for narrower columns (4,8), as it is exceedingly difficult to pack narrow-bore columns with high reduced plate heights. Figure 1 also shows the effect of system dispersion or extracolumn band-broadening as lower column efficiencies (N) are observed for early peaks (for example, the first peak, which is toluene). Note that the extracolumn effect is more severe for 2.1-mm i.d. columns because of the smaller peak volumes (6,8).



Figure 1: Comparative chromatograms of efficiency performance of three 50-mm long, 1.8-µm dp C18 columns of various inner diameters: (a) 2.1 mm, (b) 3.0 mm, and (c) 4.6 mm. Observed USP column efficiencies, N, are labelled for peaks 1, 3, and 5. An Agilent 1290 UHPLC system was used in this evaluation. Column: Agilent Zorbax Eclipse Plus C18 (50 mm, 1.8 µm); mobile phase: 70% methanol in 0.1% formic acid in water; flow rate: (a) 0.5 mL/min at 35 °C, (b) 1.0 mL/min at 35 °C, and (c) 2.0 mL/min at 35 °C; detection: 250 nm at 80 points/s; pressure: (a) 570 bar, (b) 460 bar, (c) 520 bar; sample: 1.0 µL of test mix containing (in order of peak appearance) toluene, ethylbenzene, propylbenzene, tert-butylbenzene, and anthracene.
For most users, a strong case can be made for 3-mm i.d. columns, particularly in QC applications. These columns generally have higher column efficiencies in comparison to their 2.1-mm i.d. counterparts and support practical flow rates of 0.6–1.5 mL/min. Their use may provide easier transitions for HPLC users familiar with 4.6-mm i.d. columns (15).
Gold-plated fittings with double ferrules are needed in UHPLC: Reliable fittings for UHPLC column connections and leak-free operation at high pressures were found to be problematic during the early days of UHPLC. Gold-plated nuts (to prevent seizing of the threads) and double ferrules were used in first-generation UHPLC fittings. They have fixed insertion depths and are not universally compatible with columns from different manufacturers. Currently, many choices are available, including reusable fittings for finger-tight or wrench-tight operation that can be resealed many times with a pressure rating up to 20,000 psi. Some examples found to be convenient and reliable are Opti-Lok from Optimize Technologies, VHP-320 from Idex-Upchurch, and Viper from Thermo Fisher/Dionex, though these fittings remain quite expensive. As a result of these current offerings, gold-plated nuts and double metallic ferrules are no longer requirements for UHPLC.
A binary high-pressure mixing pump is a "must": Low dwell volume is advantageous to reduce gradient delay time for fast gradients (6–8). Binary high-pressure mixing pumps have inherently low dwell volumes (because different mobile phases are pumped by two different pumps and mixing is external to the pumps). They are preferred for high-throughput screening and LC–MS applications. It was quickly found that some mixing volumes, provided by external mixers, are needed for efficient solvent blending to reduce baseline perturbations in UV detection (7,11). The optimum mixing volumes required for high-sensitivity UV detection tend to be vendor-specific and are dictated by pump designs (piston volume, availability of variable stroke volume) and the mixer type. Quaternary low-pressure mixing pumps have larger dwell volumes (because mobile phases are selected by a proportionating valve at low pressure and pumped by a single pump with mixing occurring inside the pump). They are particularly useful for method development. They also have substantially lower price tags because only a single pump is needed to form gradient or for mobile phase blending. Quaternary UHPLC pumps are now available from all major manufacturers and many have dwell volumes less than 0.5 mL, which are acceptable for most analyses by UHPLC.
UHPLC provides substantially higher UV sensitivity than HPLC: Reports on higher UV detection sensitivity with UHPLC versus conventional HPLC can be misleading. UHPLC using small internal diameter columns have often been reported to have much higher sensitivity (peak heights). This is because peak volumes are proportional to column void volumes, so a smaller column will produce a much higher peak height for the same sample amount injected. However, when the sample amount is scaled to the column volumes, both HPLC and UHPLC should yield similar sensitivity, provided detector noise and flow-cell pathlengths are equivalent. This is borne out by comparative chromatograms shown in Figure 2 of a sample analyzed on a HPLC system and an UHPLC system using identical columns with a conventional HPLC method. Detailed analysis shows the signal-to-noise ratio (S/N) and gradient shifts to be comparable on both systems. The operating pressures were 160 and 200 bar, respectively, a reflection of the smaller internal diameter connection tubing of the UHPLC system. Retention times on the UHPLC system were 0.8 to 1.4 min lower because of its smaller dwell volumes. More discussion on how to mitigate this issue during method transfers can be found in the next section. Note that an early eluted peak (for example, M235) has ~30% higher sensitivity (19 mAU in UHPLC versus 14 mAU in HPLC). This is a result of the lower dwell volume and system dispersion of the UHPLC system.

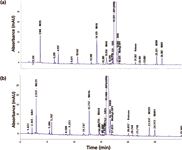
Figure 2: Comparative chromatograms of a retention marker solution for a multi-chiral drug spiked with expected impurities on (a) an HPLC (Agilent 1200 with a quaternary pump) and (b) an UHPLC system (Agilent 1290 with a binary pump). Column: 150 mm à 4.6 mm, 3.0-µm dp ACE-3-C18; mobile-phase A: 20 mM ammonium formate, pH 3.7; mobile-phase B: acetonitrile with 0.5% formic acid; gradient: 5â15% B in 5 min, 15â40% in 25 min, 40â90%B in 3 min, total run time = 42 min; flow rate: 1.0 mL/min at 30 °C; detection: 280 nm; sample: 10 µL of a retention time marker test mix containing the drug substance at 0.5 mg/mL spiked with expected impurities. Note that the noise and gradient shift were found to be comparable for the two chromatograms. The operating pressure was found to be at 160 and 200 bar, respectively. The retention times for the sample components for the UHPLC system were found to be 0.8 to 1.4 min lower than those from the HPLC system because of the lower system dwell volume (0.3 mL versus 1.0 mL).
In UHPLC, innovative flow cell designs with total internal reflectance allow the construction of smaller-volume flow cells (0.5–2 µL) with the same 10-mm pathlength as a standard HPLC flow cell (8–10 µL). This design concept has also led to the development of extended-pathlength flow cells (such as 25–60 mm) to enhance detector sensitivity (6,23). Figure 3 shows comparative chromatograms of the same sample injected on an UHPLC system with a standard (10-mm) flow cell (Figure 3[a]) and on the same UHPLC system with an extended-pathlength flow cell (60-mm) (Figure 3[b]). Note that while the noise was found to be similar (ASTM noise of 25 µAU), the signals (peak heights) were six times higher on the extended flow cell, as was expected. However, these extended-pathlength flow cells may be less useful for impurity analysis using area percent calculations because detector signal saturation can easily occur on the main peak (8). They also have higher dispersion and are therefore more compatible with larger internal diameter columns. Nevertheless, they can be advantageous for impurities testing based on external standardization, determination of trace genotoxic impurities (24), and cleaning verification applications of highly potent compounds (25).


Figure 3: Comparative chromatograms of a retention marker solution for a multi-chiral drug spiked with expected impurities on (a) an UHPLC system with a standard 10-mm UV flow cell (Agilent 1290 with a binary pump) and (b) the same UHPLC system with an extended-pathlength flow cell (60 mm). HPLC method conditions are identical to those in Figure 2 except that the injection volume is 2 µL. An ASTM noise of 24 µAU was found for both chromatograms. The limits of quantitation (LOQ) were found to be 0.05% and 0.01% for (a) and (b), respectively, at 2 µL injection. Note that an LOQ of 0.002% was found for (b) using a 10-µL injection though the main peak would saturate the detector signal.
Method transfer between UHPLC and HPLC is "a piece of cake" and method revalidation is unnecessary: Method transfer is the formal process of demonstrating that a validated method, developed or validated in one laboratory, can be properly executed by another laboratory operating under a good manufacturing practice (GMP) environment. This ensures that accurate, quality data can be generated in the latter (21). Formal method transfer between two different HPLC systems is typically not needed unless they are deemed "not equivalent." There are three scenarios for "method transfers" between HPLC and UHPLC: Same HPLC method on different types of equipment (HPLC versus UHPLC); newly developed UHPLC methods "back transfer" to HPLC conditions; and existing (legacy) HPLC methods to UHPLC methods.
Same HPLC methods on different types of equipment (HPLC and UHPLC, the simplest case): For laboratories having both HPLC and UHPLC equipment, it would be ideal if equivalent results using the same HPLC method could be obtained on both types of equipment. As demonstrated in the previous section, results are fairly equivalent with the exception of retention time shifts because of the smaller dwell volumes of a typical UHPLC (~0.3 mL for UHPLC versus ~1.0 mL for HPLC). This can be remedied by several means: Increasing the dwell volume of UHPLC system by using a larger external mixer (probably not very practical); building an initial isocratic segment into the HPLC method and allowing the user to adjust the duration of this segment in the method (generally preferred); or using optional simulation software available on some chromatography data systems to simulate the performance of various equipment by automatic method adjustments (26,27).
"Back transferring" or "translating" UHPLC methods to HPLC method conditions (in method development labs): Many laboratories prefer to use UHPLC for rapid method development including column and mobile phase screening and method optimization (5–7,21) and then "back transfer" the optimized UHPLC methods to HPLC conditions using longer column with larger particles via geometrical scaling. The approach is typically used to support global manufacturing operations since UHPLC may not be available universally. Case studies for method transfer processes are available elsewhere (7,21,28,29).
Method transfer from HPLC to UHPLC (for GMP operation): The primary driver to purchase UHPLC equipment is the ability to perform faster analysis with "good" resolution. A 2–3-fold or greater reduction in analysis time, while maintaining similar resolution, is readily achievable using UHPLC. For instance, a 15-min method using a 150 mm × 4.6 mm column packed with 5-µm particles can theoretically be performed on a 50 mm × 2.1 mm column packed with 1.7-µm particles with equivalent column efficiency in 5 min (10). Even faster analysis (1.5 min or a ninefold increase) is possible if optimum flow rates are used (optimum linear velocity is inversely proportional to particle size). A geometrical scaling approach is typically used to accomplish such transfers (29).
Some ground rules for method scaling between HPLC and UHPLC: Column length is scaled to particle size keeping the column length to particle size ratio the same; flow rate is scaled to cross-sectional area of the column (also inversely proportional to the particle size if optimum flow can be used); gradient time is scaled to column length; and flow rate and injection volume are scaled to column void volume. One important requirement is that the new UHPLC column used must contain identical bonded phase materials to eliminate any selectivity differences. Also, mobile phases used should be identical (type of buffer, strength, pH, and organic modifier). Details on this geometrical scaling are available from the Pharmacopeial Forum (29) and calculator programs are available at various vendors' websites (Waters, Agilent, and Thermo Fisher/Dionex) and other sources (30).
For validated HPLC methods, there were numerous discussions on what constitutes a method adjustment versus a method change, and at what point a method revalidation is needed (21,31). The current consensus appears to be that a partial method validation (including specificity, intermediate precision, linearity, and robustness) should be considered as well as a demonstration of method equivalency between the two methods (7,21,28,31). This process may be straightforward for simple assays but can be challenging for complex samples (15,21), particularly for QC methods for commercial products.
Lower-dispersion UHPLC systems are better — some pros and cons: This statement may not be a myth because lower system dispersion is always considered to be better (6,8). Low system dispersion systems are desirable because they allow the use of smaller columns without efficiency loss. However, there are also some important caveats and trade-offs. Lower dispersion is achieved by a reduction of the volume of sample fluidic path (that is, sample loop, switching valve, connection tubing, and flow cell). Table 1 shows a compilation of comparative system dispersion measurements (5σ bandspread or instrumental bandwidth) of a number of HPLC and UHPLC systems (6,32). It should be noted that UHPLC systems have substantially lower dispersion than convention HPLC systems and larger sample loop or flow cell, switching valve, and connection tubing all contribute to system dispersion or bandwidth.

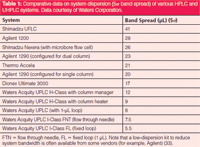
Table 1: Comparative data on system dispersion (5Ï band spread) of various HPLC and UHPLC systems. Data courtesy of Waters Corporation.
It is useful to realize that system dispersion before the column (injector, loop, switching valve) is generally less important since most high-resolution analyses are conducted under gradient conditions (since sample bands are refocused at the top of the column). Post-column dispersion (tubing from column to detector and detector flow cell or mass spectrometer source) is more critical because it will broaden separated bands. Nevertheless, post-column tubing and detector UV flow cells can easily be changed in some cases. Note that a low-dispersion kit to reduce system bandwidth is often available from some vendors (such as Agilent) (33). Also, the use of small sample loops (<20 µL), column ovens (<200 mm), and connection tubing (<0.003 in. i.d. which generates substantial back pressure at flow rates greater than 1 mL/min) in some low-dispersion systems, can be less compatible with legacy HPLC methods. So, lower system dispersion is a good thing for demanding applications for maximum performance — but may lead to some sacrifice in system convenience and flexibility for routine analysis with diverse methods.
Summary and Conclusions
This instalment addresses eight popular myths in UHPLC and provides evidence and references to delineate and repudiate some of these beliefs. Here is a summary of the conclusions:
- UHPLC is complementary to high-temperature LC and core–shell columns and can be used by itself or in combination with these approaches.
- Viscous heating is not a "huge" practical issue for sub-2-µm particle columns using still-air ovens under "normal" operating conditions.
- A 2.1-mm i.d., sub-2-µm column is a common column for UHPLC; however, a strong case can be made for 3-mm i.d. columns, particularly for QC applications.
- There are many excellent fittings that can be used and resealed many times with pressures up to 20,000 psi. Gold-plated nuts and double ferrules are not a requirement.
- A binary high-pressure mixing pump is preferred for high-throughput separations, though most major vendors also offer quaternary low-pressure mixing pumps with marginal increases in dwell volumes.
- UHPLC systems do not provide substantially higher sensitivity in UV detection with standard 10-mm long UV flow cells. However, 2–6-fold increases are possible with the use of extended-pathlength flow cells (25 to 60 mm long).
- Method transfer (translation) between UHPLC and HPLC can be challenging for complex methods. A partial method revalidation is a typical regulatory expectation (or requirement) including a demonstration of method equivalency.
- Lower-dispersion UHPLC systems are indeed better but expect some sacrifice in flexibility with respect to injection volumes, compatibility to longer columns, and higher flow rates required by routine analysis with legacy HPLC methods.
Acknowledgements
The author is grateful to Sam Yang, Christine Gu, Mohammad Al-Sayeh, and Eileen Zhao of Genentech; Jim Jorgenson of the University of North Carolina; Davy Guillarme and Szabolcs Fekete of the University of Geneva; Raphael Ornaf of Vertex Pharmaceuticals; Ken Broeckhoven of Vrije Universiteit Brussel; Tom Waeghe of MacMod; John Dolan from LC Resources; Bill Barber from Agilent Technologies; Pam Iraneta and Eric Grumbach of Waters; Joe DiCesare and Wilhad Reuter of PerkinElmer; and Ross Woods of the University of Texas at Arlington. It should be recognized that the design of modern UHPLC equipment constitutes many trade-offs between system bandwidth, sensitivity, and compatibility to conventional HPLC methods, dwell volume, mixing efficiency (UV sensitivity), and cost, flexibility, and reliability. The data collected here stemmed from equipment and columns available at the time of evaluation and may not be representative of those currently available. The opinions expressed in this article are solely those of the author and bear no reflection on those of LCGC Asia Pacific or other organizations.
Michael W. Dong is a senior scientist in Small Molecule Drug Discovery at Genentech in South San Francisco, California, USA. He is responsible for new technologies, automation and supporting late-stage research projects in small molecule analytical chemistry and QC of small molecule pharmaceutical sciences. He holds a PhD in analytical chemistry from the City University of New York, USA, and a certificate in Biotechnology from U.C. Santa Cruz, USA. He has conducted numerous courses on HPLC/UHPLC, pharmaceutical analysis, HPLC method development, drug development process and drug quality fundamentals. He is the author of Modern HPLC for Practicing Scientists and a co-editor of Handbook of Pharmaceutical Analysis by HPLC. He is a member of the editorial advisory board of LCGC North America.
References
(1) B.A. Bidlingmeyer, R.P. Hooker, C.H. Lochmuller, and L.B. Rogers, J. Sep. Sci. 4(6), 439–446 (1969).
(2) J.E. MacNair, K.C. Lewis, and J.W. Jorgenson, Anal. Chem. 69, 983–989 (1997).
(3) N. Wu, J.A. Lippert, and M.L. Lee, J. Chromatogr. A 911, 1–12 (2001).
(4) U.D. Neue, M. Kele, B. Bunner, A. Kromidas, T. Dourdeville, J.R. Mazzeo, E.S. Grumbach, S. Serpa, T.E. Wheat, P. Hong, and M. Gilar, in Advances in Chromatogr., S. Fanali, P.R. Haddad, C. Poole, P. Schoenmakers, and D.K. Lloyd, Eds. (Elsevier/CRC Press, Boca Raton, Florida, USA, 2009), pp. 99–143.
(5) D. Guillarme, J.-L. Veuthey, and R.M Smith (Ed), UHPLC in Life Sciences (Royal Society of Chemistry Publishing, Cambridge, United Kingdom, 2012).
(6) K.J. Fountain and P.C. Iraneta, in UHPLC in Life Sciences, D. Guillarme, J.-L. Veuthey, and R.M Smith, Eds. (Royal Society of Chemistry Publishing, Cambridge, United Kingdom, 2012), pp. 283–311.
(7) M.W. Dong, LCGC North Am. 25(7), 656–666 (2007).
(8) M.W. Dong, Modern HPLC for Practicing Scientists (Wiley, Hoboken, New Jersey, USA, 2006).
(9) N. Wu and A.M. Clausen, J. Sep. Sci. 30, 1167–1182 (2007).
(10) D. Guillarme and M.W. Dong, Amer. Pharm. Rev. 16(4), 36–43 (2013).
(11) M.W. Dong, in Chromatography: A Science of Discovery, R.L. Wixom and C.W. Gehrke, Eds. (Wiley, Hoboken, New Jersey, USA, 2010), pp. 328–332.
(12) D.T.T. Nguyen, D. Guillarme, S. Rudaz, and J.L. Veuthey, J. Sep. Sci. 29, 1836–1848 (2006).
(13) D. Guillarme, J. Ruta, S. Rudaz, and J.-L. Veuthey, Anal. Bioanal. Chem. 397, 1069–1082 (2010).
(14) D. Guillarme, E. Grata, G. Glauser, J.-L. Wolfender, J.-L. Veuthey, and S. Rudaz, J. Chromatogr. A 1216, 3232–3243 (2009).
(15) M.W. Dong, D. Guillarme, S. Fekete, R. Rangelova, J. Richards, D. Prudhomme, and N.P. Chetwyn, J. Pharm. Biomed. Anal. submitted.
(16) J. Ruta, D. Guillarme, S. Rudaz, and J.L. Veuthey, J. Sep. Sci. 33, 2465–2477 (2010).
(17) S. Heinisch, in UHPLC in Life Sciences, D. Guillarme, J.-L. Veuthey, and R.M. Smith, Eds. (Royal Society of Chemistry Publishing, Cambridge, United Kingdom, 2012), pp. 102–128.
(18) S. Fekete, E. Oláh, and J. Fekete, J. Chromatogr. A 1228, 57–71 (2012).
(19) L. Nováková, J.-L. Veuthey, and D. Guillarme, J. Chromatogr. A 1218, 7971–7981 (2011).
(20) F. Gritti and G. Guiochon, Anal. Chem. 80, 5009–5020 (2008).
(21) B. Debrus, E. Rozet, P. Hubert, J.-L. Veuthey, S. Rudaz, and D Guillarme in UHPLC in Life Sciences, D. Guillarme, J.-L. Veuthey, and R.M. Smith, Eds. (Royal Society of Chemistry Publishing, Cambridge, United Kingdom, 2012), pp. 67–98.
(22) K. Broeckhovn, J. Billen, M. Verstraeten, K. Choikhet, M. Dittmann, G. Rozing, and G. Desmet, J. Chromatogr. A 1217, 2022–2031 (2010).
(23) A. Gratzfeld-Huesgen, Agilent Technologies, 2012, 5991-0115EN.
(24) A. Teasdale, Ed., Genotoxic Impurities: Strategies for Identification and Control (Wiley, Hoboken, New Jersey, USA, 2011).
(25) M.W. Dong, E.X. Zhao, D.T. Yazzie, C.C. Gu, and J.D. Pellett, Amer. Pharm. Rev. 15(6), 10–17 (2012).
(26) M. Dittmann, K. Choikhet, P. Stemer, and K. Witt, "Method Transfer Between UHPLC and HPLC: Issues and Solutions," presented at Pittcon 2011, Atlanta, Georgia, USA, 2011.
(27) Agilent 1290 Infinity LC with Intelligent System Emulation Technology, Agilent Technologies, 20135990-8670EN, 2013.
(28) G. Vanhoenacker, F. David, P. Sandra, B. Glatz, and E. Naegele, Agilent Applications Notes, 5990-3981 EN, 2009.
(29) U.D. Neue, D. McCabe, V. Ramesh, H. Pappa, and J. DeMuth, Pharmacopeial Forum 35(6), 1622–1626, 2009.
(30) D. Guillarme, http://www.unige.ch/sciences/pharm/fanal/lcap/telechargement-en.htm.
(31) M. Swartz and I. Krull, LCGC North Am. 24(8), 480–490 (2006).
(32) S. Fekete, I. Kohler, S. Rudaz, and D. Guillarme, J. Pharm. Biomed. Anal., in press, http://dx.doi.org/10.1016/j.jpba.2013.03.012.
(33) J.J. Stankovich, F. Gritti, P.G. Stevenson, and G. Guichon, J. Sep. Sci. 36(17), 2709–2717 (2013).











Removing Double-Stranded RNA Impurities Using Chromatography
April 8th 2025Researchers from Agency for Science, Technology and Research in Singapore recently published a review article exploring how chromatography can be used to remove double-stranded RNA impurities during mRNA therapeutics production.


The Effect of Time and Tide On PFAS Concentrations in Estuaries
April 8th 2025Oliver Jones and Navneet Singh from RMIT University, Melbourne, Australia discuss a recent study they conducted to investigate the relationship between tidal cycles and PFAS concentrations in estuarine systems, and offer practical advice on the sample preparation and LC–MS/MS techniques they used to achieve the best results.



