Metal-Analyte Interactions—An Unwanted Distraction
The interaction of transition metals with analytes, present at low concentrations in samples, has been a problem for analytical scientists for many years. Such unwanted interactions often result in poor peak shapes, loss of sensitivity, and, in some instances, truncated linear dynamic ranges. Whilst a number of pragmatic solutions to this problem have been advanced, all suffer from practical difficulties that limit their application. Recently a new method to deal with this problem has been developed that shows promise for greatly attenuating, or even eliminating, such adverse surface interactions with minimal effects on the routine operation of liquid chromatography (LC) systems.
The wicked sense of humour that medicinal chemists have is never more apparent than when they send their favourite compounds—having had them dosed to their in vivo model of choice—for bioanalysis. This is often because, to their surprise, these candidates are not as effective in vivo as they had expected based on their zeptomolar in vitro potency. The unsuspecting bioanalyst is then confronted with molecules offering a surprising array of problems that they have to solve in order to develop a sensitive and specific method for this new (insoluble, highly hydrophilic/hydrophobic/amphoteric/high-molecular-weight [HMW]/low MW) compound, and then return reliable, quantitative data with a rapid turnround.
The sample volumes supplied are often quite small as the adoption of microsampling, as part of replace-reduce-refine strategies for animal use, continues and this further complicates matters because it results in low absolute masses of analyte available for bioanalysis. If the drug candidates in such investigations have a tendency towards non-specific binding or adsorption onto the component parts of the LC system or column, then the loss of the already low concentration analyte is likely. These losses have clear and obvious adverse effects on accurate determination of analyte concentrations and consequences for the correct calculation of factors such as the pharmacokinetics/dynamics (PK/PD) of the analyte. Indeed, such effects may preclude the proper characterization of the elimination phase of candidate drugs and, if there are similar effects on their metabolites, may confound efforts to characterize other drug metabolism and pharmacokinetic (DMPK) aspects of the study.
Whilst interaction with silanols on the silica‑based stationary phases commonly used for LC are widely known and appreciated—and now widely mitigated—a further, less common, source of analyte-specific adsorption can be the result of interactions with any transition metals present in the LC systems, tubing, and columns. Acting as Lewis acids, these metals interact with those analytes containing functional groups, such as phosphates or various types of amine, hydroxy, and carboxylic acid that can chelate metals. The outcome of these interactions is poor, sometimes unacceptable, chromatographic peak shape and the complete loss of the target analyte, particularly at low concentration. A range of strategies have been tried to reduce or eliminate such unwanted analyte-system interactions including buffers, additives such as ion-pairing (IP) or chelating reagents, or attempts at the removal of metals from the system by constructing columns using PEEK instead of traditional metals, such as stainless steel. These strategies can provide a measure of success but are not routine approaches, and whilst they are able to reduce analyte-metal interactions they may have detrimental effects on sensitivity, with mobile phase additives competing for mass spectrometry (MS) ionization. PEEK also has limited mechanical strength and is only suitable for relatively low pressure operation, constraining its use in ultrahigh-pressure liquid chromatography (UHPLC) systems. Whilst it is possible to line columns with PEEK to overcome the pressure constraints, there can still be problems with solvent compatibility (for example, DMSO) and this does not address the interactions with frits and the other metal components of the LC system that may come into contact with the analyte. Another approach to “passivating” the system is to perform repeated injections of the analytes until the signal stabilizes, but, whilst partially effective, this “solution” does leave the analyst feeling a little uncomfortable about the accuracy and robustness of the method. This leaves adding chelating agents to the mobile phase (EDTA, or picolinic acid), which is probably acceptable for LC–UV‑based methods but, depending upon the concentration required, may cause problems for LC–MS and be a source of long-term contamination.
The ideal answer is to eliminate the problem entirely by masking the “active sites” in the LC system and column, thereby rendering them inaccessible to the analytes. A novel hybrid surface technology (HST) surface barrier (1) does just this, having been conceived and developed to address such problems, by forming an effective barrier that prevents the metal surfaces and analyte molecules from reaching each other. The concept is based on the use of a highly cross-linked surface treatment, similar to the organo-silica hybrid chemistries used to form silica-based UHPLC stationary phases. The surface that is formed provides an inert and resilient barrier, blocking access to the metal surfaces but not participating in the ongoing separations.


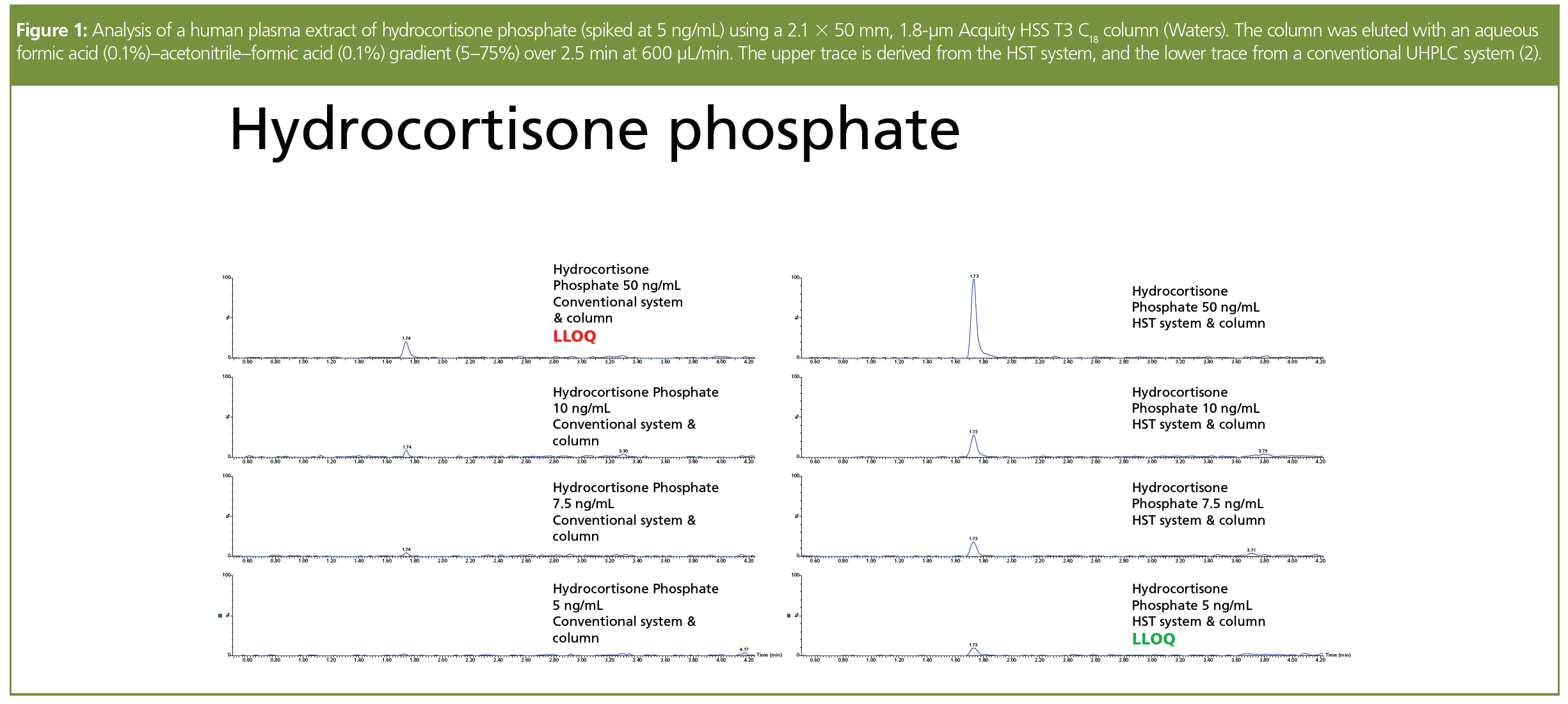
Using this strategy has a number of benefits, such as eliminating the need for mobile phase additives whilst improving peak shape and increasing recovery from the column (all of which contribute to improved sensitivity). It also eases method development for these “metalophilic” analytes as this becomes no more difficult than for most “normal” compounds. A good example of the improvements that can be obtained is seen with phosphorus-containing prodrugs, such as the steroids dexamethasone and hydrocortisone. These anti-inflammatory corticosteroids are widely prescribed for treating endocrine disorders, autoimmune, and allergic conditions including arthritis, psoriasis, lupus, and ulcerative colitis. More recently, dexamethasone phosphate has been shown to be a cheap and effective means of treating the respiratory symptoms of COVID-19 patients. In Figure 1, the improvement in signal response for a plasma extract of hydrocortisone phosphate (prepared by protein precipitation) on an HST system, operated in reversed-phase mode, compared to a conventional system is impressive (2). In the case of the HST method, the surface treatment was applied to the full range of metal surfaces that analytes could come into contact with (that is, the metal surfaces within the flow path of the LC system, column frits, and the walls of the chromatographic column). Similar improvements were seen for dexamethasone phosphate. For both of the steroid prodrugs it was interesting to note that at higher concentrations (100 ng/mL), the MS detector response produced was similar for both the HST and conventional UHPLC systems. However, as the analyte concentration was reduced a marked difference in peak response was observed between the two systems. These observations suggest that at low analyte (on-column) mass loadings the steroid phosphate was completely adsorbed on the metal surfaces of the conventional LC system but that this did not occur within the HST system, enabling the analyte’s detection. At higher analyte concentrations the same mass of analyte material interacted with the metal surfaces in the conventional LC system as for the low concentration sample, with the majority of the analyte passing to the mass spectrometer for detection. The analysis of a large batch of steroid-phosphate prodrug-containing plasma samples on a conventional LC system showed that, despite making greater than 100 injections of plasma extract to passivate the active sites, the signal response for the low concentration sample (5 ng/mL) never approached that obtained with the HST system. This demonstrates that the aim of system passivation, which would allow parity in performance to be achieved, was not achieved to any significant level. The derived statistical data from the HST system, produced during assay validation, showed a reduced %CV not only for the low concentration standards/quality controls (QCs) but also for those of medium concentration, thus simplifying method development and improving performance (2).
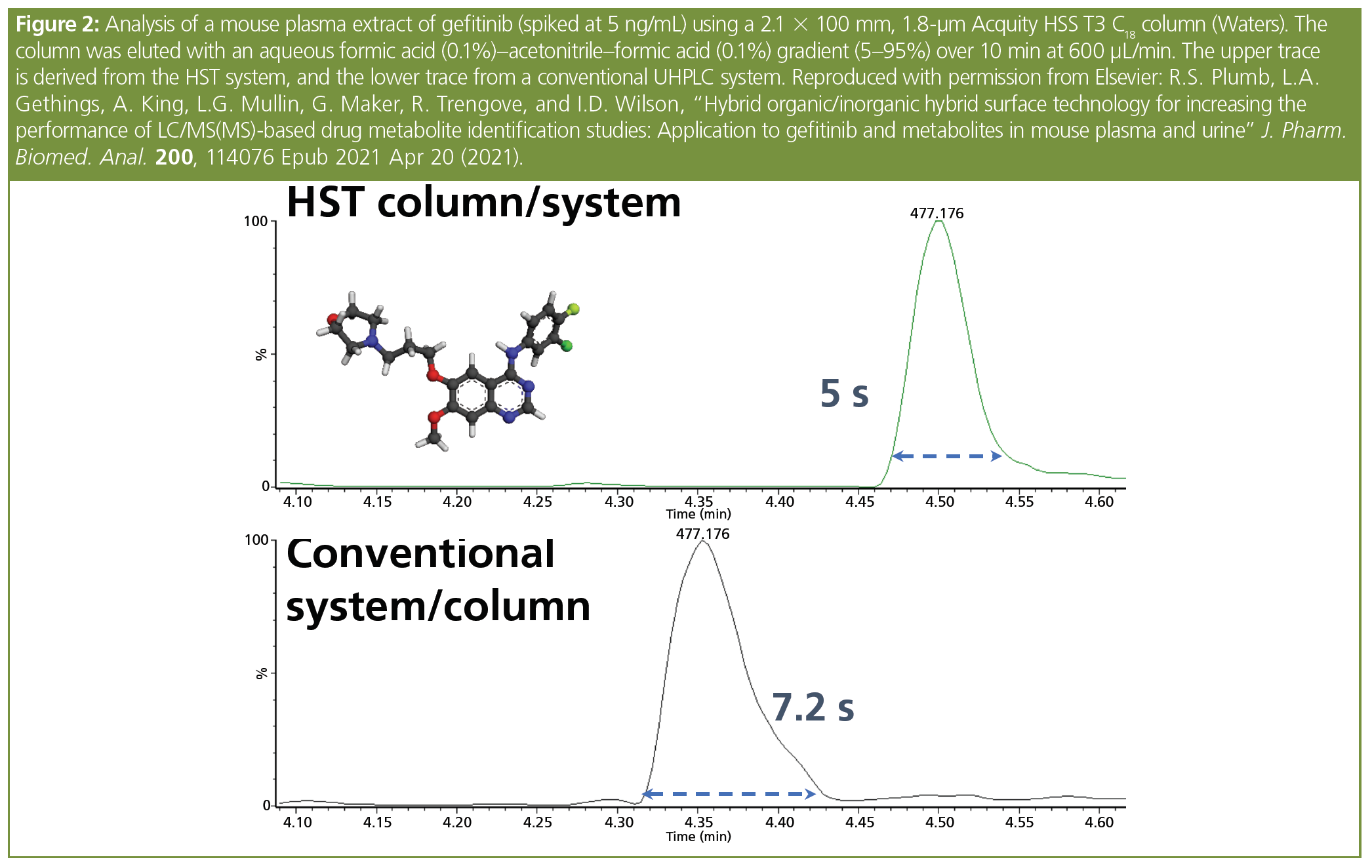
As outlined above, the interaction of many analytes containing phosphates, various types of amines, hydroxy, and carboxylic acid groups with transition metals in LC systems has been previously reported. Thus, any improvements in materials science that mitigate these Lewis acid-base interactions would show a benefit in the analysis of phosphate-containing analytes, such as the steroid prodrugs illustrated here. Whilst the majority of candidate pharmaceuticals do not contain these active groups, many still exhibit peak tailing, which requires the addition of buffers to the mobile phase to improve the chromatographic peak shape. One such compound is the EGFR inhibitor gefitinib, a drug employed for treating certain breast, lung, and other cancers. For conventional reversed-phase LC, ammonium acetate has to be used in the eluent to obtain suitably symmetrical peaks. Although the molecule is not an obvious metal chelator, a comparison of HST and standard UHPLC systems for the analysis of gefitinib and metabolites in mouse urine and extracts of plasma obtained after a 50 mg/kg oral dose demonstrated clear improvements in both peak shape and intensity. For gefitinib itself, peak tailing was reduced by a third and peak response increased by approximately 70% (see Figure 2); similar improvements were seen for some but not all metabolites (3).

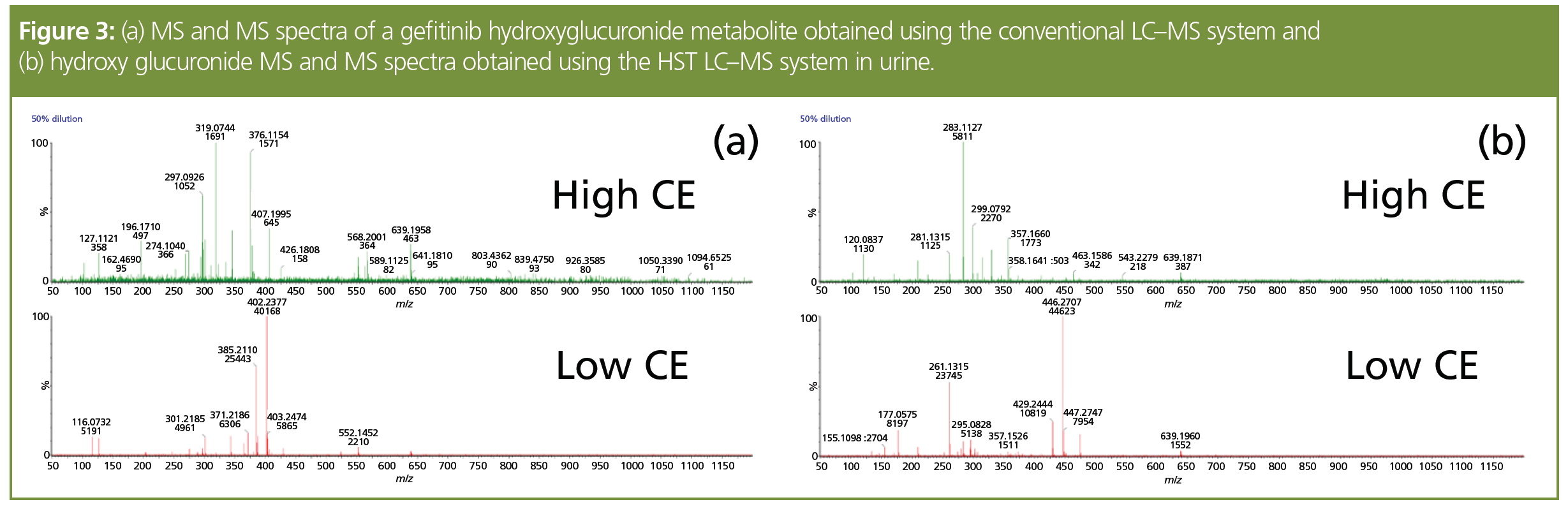
These improvements in LC performance produced by the HST system for the quantitative analysis of pharmaceuticals in biofluids were not limited to peak shape and intensity but were also evident in the enhanced quality MS and MS/MS data obtained for structural elucidation of metabolites. So, in the case of the gefitinib metabolites detected in pooled mouse urine, a comparison of conventional vs. HST UHPLC systems highlighted a clear improvement in both the intensity of the MS data obtained and the quality of spectra. This advantage is clearly illustrated in Figure 3, which shows the MS and MS/MS data generated on a hydroxyglucuronide metabolite of gefitinib. From the low energy MS spectra it is clear that the precursor ion m/z 639.196 is obvious in the HST data but absent from the spectrum from the conventional
UHPLC–MS analysis. In the high collision energy data, the diagnostic fragment ion at m/z 463.152 is easily observed and interpreted as opposed to the conventional UHPLC data where this ion is not detected and the spectrum also contains many fragment peaks, unrelated to the drugmetabolite, arising from the incomplete resolution of the target metabolite from endogenous contaminants in the samples/matrix.

Successful bioanalysis has certain non‑negotiable requirements. A method must provide sufficient sensitivity and linear dynamic range to determine the pharmacokinetic elimination phase. The LC efficiency has to resolve the analytes of interest from interferences (endogenous or drug-related) to provide specificity. Critically, it must be sufficiently robust and reproducible to allow routine acquisition of data from large batches of samples.
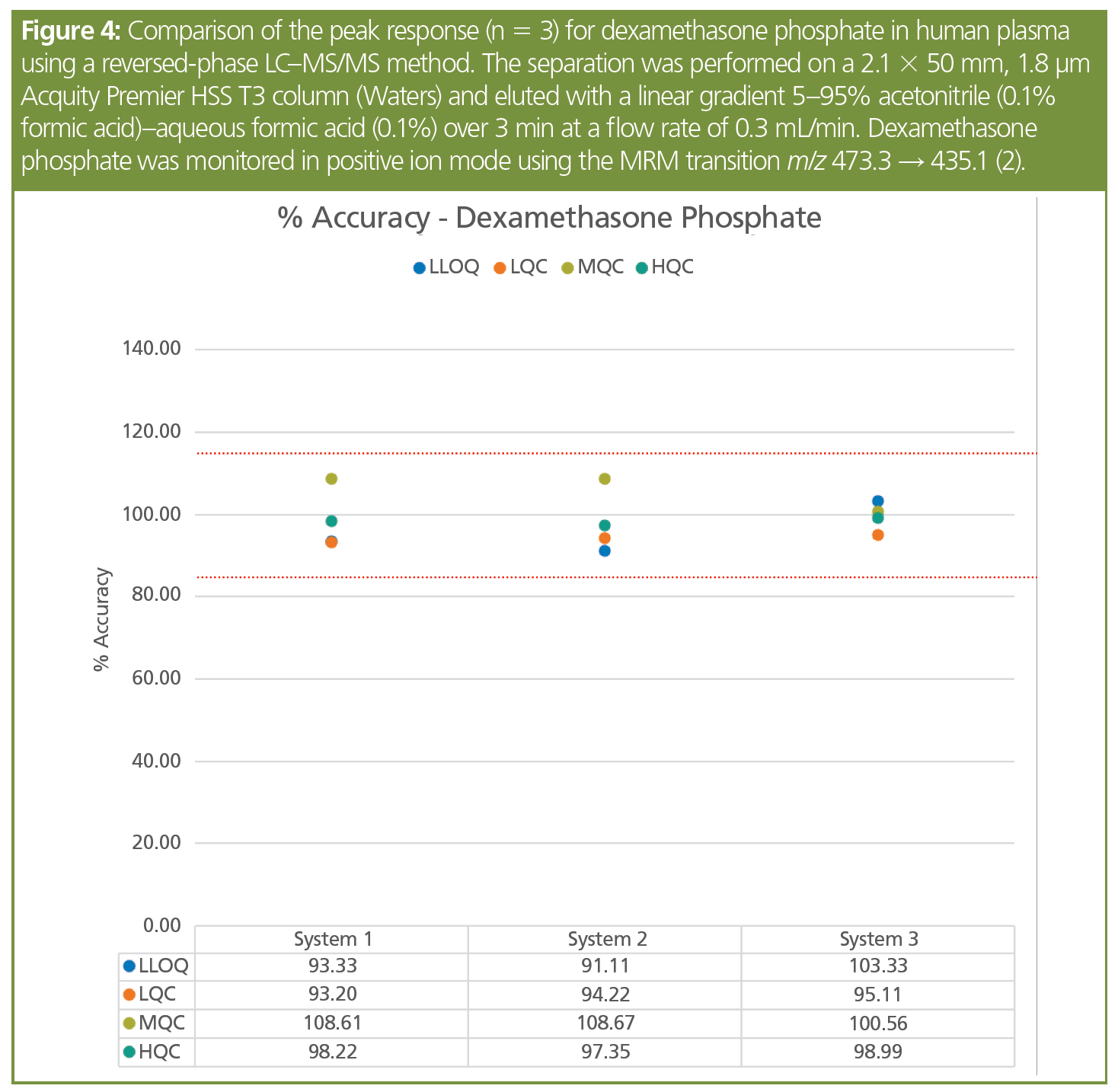
Method migration and transfer between instruments and laboratories should ideally also be straightforward. However, instrument‑to-instrument variability often makes this process both tedious and labour‑intensive, with problems that frequently manifest themselves including differences in analyte retention, peak shape, and limits of detection. Some of these can be laid at the door of analyte adsorption onto the different components of the LC system (for example, different lengths/volumes of connection tubing or differences in the materials employed). The inert surface barrier technology of the HST helps to eliminate these adsorption-related variances in performance. This is illustrated in Figure 4 where the accuracy of the HST LC–MS/MS system is shown for method transfer between three separate systems at the lower limit of quantification (LLOQ), limit of quantification (LOQ), mid-range QC (MQC), and high QC (HQC) for dexamethasone phosphate. As can be seen from these data, the analysis of the samples for all of the concentrations examined was consistent across the three systems over three separate occasions.

Conclusion
The HST method described here provides a convenient and readily implemented solution to many of the problems associated with analytes that can interact with metals in conventional LC systems and columns. These results did not require the use of mobile phase additives that might otherwise adversely affect MS performance and did not require system passivation (saving
both time and costly reagents). In addition, the benefits of the HST are achieved
without detriment to analytes that are not prone to metal-sensitive adsorption in the first place.
References
- M. DeLano, T.H. Walter, M.A. Lauber, M. Gilar, M.C. Jung, J.M. Nguyen, C. Boissel, A. Patel, A. Bates-Harrison, and K.D. Wyndham, Anal. Chem. 93(14), 5773–5781 (2021).
- N. Tanna, L.G. Mullin, P.D. Rainville, I.D. Wilson, and R.S. Plumb, J. Chromat. B 1179, 122825 (2021).
- R.S. Plumb, L.A. Gethings, A. King, L.G. Mullin, G. Maker, R. Trengove, and I.D. Wilson, J. Pharm. Biomed. Anal. 200, 114076 (2021).
Robert Plumb is the Director of Omics and Small Molecule Pharma in the Waters Scientific Operations Division in Milford, Massachusetts, USA. He is responsible for the development of new approaches to DMPK, proteomics, and metabolomics. Prior to his current position, he was at GlaxoSmithKline for 15 years in the areas of bioanalysis, drug metabolism, and metabolic profiling. He has published over 100 papers on the subject of HPLC–MS and NMR for bioanalysis, metabolomics, and metabolite identification. He is an Adjunct Professor at Murdoch University and a Fellow of the Royal Society of Chemistry. In 2014 he was awarded Highly Cited Researcher by Thompson Reuters.
Ian Wilson has worked in the areas of bioanalysis, metabolic phenotyping, and drug metabolism in the pharmaceutical industry for over 30 years, most recently as a Senior Principal Scientist in the Department of Drug Metabolism and Pharmacokinetics at the AstraZeneca Research site at Alderley Park (Macclesfield, UK). He moved to Imperial College London in 2012 to a personal Chair in Drug Metabolism and Molecular Toxicology, where he is now a visiting Professor. Research interests are the development and application of advanced analytical techniques (especially those linked to high resolution and/or high-throughput separation methods) to problems in drug metabolism, toxicology, and metabonomics, resulting in 570 publications. He is an Editor of the Journal of Chromatography (B) and therecipient of a number of awards in chromatography and analysis, such as the Chromatographic Societies’ Martin Medal and the Knox Medal of the Royal Society of Chemistry Separation Science Group. He was recently awarded an Honorary Doctorate from the University of Thessaloniki and was made an Honorary Fellow of the Metabolomics Society.












