The LCGC Blog: Forensic Drug Analysis: GC–MS versus LC–MS
If given the need to determine drug A or its metabolite in blood, 99% of the time I would choose to start with liquid chromatography–mass spectrometry (LC–MS).


kwanchaift/stock.adobe.com

If given the need to determine drug A or its metabolite in blood, 99% of the time I would choose to start with liquid chromatography–mass spectrometry (LC–MS).
When I asked a couple of former students who work in a forensics crime laboratory the same question, they said they were split about 50/50 on the methods they use for drugs of abuse that feature liquid chromatography–mass spectrometry (LC–MS) versus gas chromatography (GC)–MS. It was actually a relief to hear that there was only a 50% skew towards GC–MS. For the past two years, my group has run a blood drug analysis course at UT-Arlington for defense lawyers through the National College for DUI Defense (NCDD). During the courses, I gave an introduction to LC–MS. Last year, not a single lawyer attending the course had previously come across LC–MS forensics analysis in the courtroom. In fact, the vast majority had not heard of liquid chromatography before. This year, two out of 24 lawyers in the course indicated they had heard about LC–MS, but they had still not faced such evidence in a case.
Anecdotally, it sounds like there is not a lot of variation in forensic drug analysis across the country. My students in their crime laboratory appear to be in the minority (and ahead of the curve). One lawyer in our course indicated that in the state they practice, their laboratories use GC–flame ionization detection (GC–FID) for blood drug analysis. If that does not make you worry, then it should. I am so flabbergasted by this, I don’t even want to mention the state-luckily, it is not a heavily populated one.
GC–FID is much less sensitive and specific compared to GC–MS. In combination with headspace sampling, GC–FID is still used to a great degree for blood alcohol content (BAC) determinations. BAC measurements do not require a great deal of sensitivity, and there are very few volatile interferences in blood. Even so, GC–MS would still be preferred. GC–FID can only identify a compound based on its retention time, whereas GC–MS also provides mass spectral information for each peak. When you get to the trace levels of drugs and metabolites in biological fluids, you need sensitivity and specificity; these are readily provided by an MS detector.
In our short course, I asserted to the lawyers that the use of LC–MS for drug analysis is on its way. Eventually, it will show up in the courtroom-but why is it taking so long to get there? It is obvious that GC–MS is a more established technique in forensics laboratories. It also appears that forensics labs are generally resistant to change. Why should they change if the way they are currently generating data is acceptable to the courts? I could easily argue that the courts do not, in general, know any better, but maybe it is instructive to make a little comparison.
Table 1 provides some points for comparison of GC–MS, for an electron ionization (EI) source and a singleâquadrupole mass analyzer, with LC–MS, using electrospray ionization (ESI) and tandem mass spectrometry (MS/MS) on a triple-quadrupole instrument.

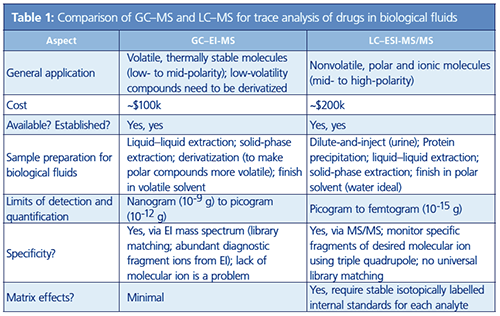
Without diving in too deeply, there are a few aspects that make LC–MS better amenable for determination of drugs in biological fluids. First and foremost, LC–MS is ideal for applications involving drugs, which generally are polar and nonvolatile molecules. Efficient separation and ion generation can be accomplished using LC–MS without derivatizing the analyte. For GC–MS, derivatization is necessary to chemically modify the drug and make it more GC-amenable (that is, more volatile). Underivatized drugs analyzed by GC–MS will generally exhibit poor peak shapes (reduced resolution) and reduced sensitivity. A derivatization step can also add significant uncertainty. In general, sample preparation steps are more prone to error than other parts of an analytical method. More sample preparation steps equate to increased error in determinations, and chemical derivatization is an intensive process that can be prone to uncertainties, such as the quality and age of reagent, presence of interferences in the sample, variability in laboratory conditions, and so on. LC–MS generally requires less sample preparation. For a urine analysis, one can often just dilute with water and inject the sample.
With LC–MS, using ESI, an intact molecular ion is generated and enters the mass spectrometer. Using GC–EI-MS, the signal for a molecular ion may not be readily apparent because of the extensive fragmentation generated by the 70-eV ionization energy. Even so, EI generates a nicely diagnostic mass spectrum that can be reproduced across different instruments, and only requires a single quadrupole mass analyzer. GC–MS is generally cheaper because it uses a less sophisticated detector. For LC–MS, one gains specificity with the use of a triple-quadrupole mass analyzer. In the triple quadrupole, the initial ion of interest is isolated, then fragmented in a collision cell, and then unique ion fragments of interest are used to quantify the compound and confirm identity.
In this MS/MS operation, signal-to-noise (S/N) levels are increased dramatically by reduction of noise. As a result, LC–MS determinations usually exhibit lower limits of quantification (LOQs) than GC–MS determinations (although this may be application specific). These lower LOQs are important for forensics drug analysis because one does not want to be operating very close to the LOQ if it can be avoided. First, determinations made closer to the LOQ are usually subject to higher error. Second, when one operates close to the LOQ, it is possible to lose some of the specificity of the determination. Confirmation of the target can be made based on the presence of three or four ions (and a consistent intensity ratio between them); this statement is true for both GC–EI-MS and LC–MS/MS. Because LC–MS is more sensitive, there is a better chance for those ion ratios to be preserved when the compound is present at low levels, especially levels close to the LOQ for GC–MS, where some lower abundant ion fragments would not be observed.
To be fair, a place where LC–MS has problems relative to GC–MS is with matrix effects. The matrix is everything else in the sample besides the analyte. In biological fluids, there are ample species in the matrix that can interfere with an analyte measurement. Matrix effects can cause unwanted changes in analyte response; they will compromise the accuracy of the determination if they are not properly accounted. For this reason, it is necessary to use stable isotopically labelled internal standards (SIL-IS) for each analyte of interest. This means that for each analyte, a pure isotopically labelled internal standard (a deuterated, 13C-labelled, or 15N-labelled version of the analyte) must be used to normalize matrix effects. This internal standard will be eluted with the analyte, but its signal will be differentiable from the analyte because it has a different mass, and it will yield different fragment ions. These compounds are expensive, but without their use, LC–MS determinations of drugs from biological fluids will be subject to significant error and largely considered unreliable.
This topic could be discussed a great deal more. I expect to address it more in the future. The interface between the forensic and legal communities is one where getting the analysis right is more important than ever. People go to jail or pay massive fines for illegal actions involving drugs. It should be the responsibility of the crime laboratories to use the best technology for their analysis, to avoid error in their case assessments. If you are still wondering whether the evidence is strong enough favouring LC–MS over GC–MS for drug analysis from biological fluids, go to any clinical laboratory and count the relative number of GC–MS and LC–MS instruments they have. GC–MS will lose. GC–MS simply cannot provide adequate performance across the large and growing range of illegal substances desired to be detected. On the contrary, one would not need to look far for demonstrations of LC–MS methodologies that can handle virtually all drug compounds of interest, using a single method with minimal sample preparation (1). LC–MS instrumentation and processes may be more complicated than GC–MS, and there might be a couple of different controls to have in place, but LC–MS should be the future of forensic drug analysis.
Reference
- S. Lupo, Restek Corporation. https://www.restek.com/pdfs/CFAR2309-UNV.pdf


Kevin A. Schug is a Full Professor and Shimadzu Distinguished Professor of Analytical Chemistry in the Department of Chemistry and Biochemistry at The University of Texas (UT) at Arlington, Texas, USA. He joined the faculty at UT Arlington in 2005 after completing a Ph.D. in chemistry at Virginia Tech under the direction of Prof. Harold M. McNair and a postdoctoral fellowship at the University of Vienna under Prof. Wolfgang Lindner. Research in the Schug group spans fundamental and applied areas of separation science and mass spectrometry. Schug was named the LCGC Emerging Leader in Chromatography in 2009 and the 2012 American Chemical Society Division of Analytical Chemistry Young Investigator in Separation Science. He is a fellow of both the U.T. Arlington and U.T. System-Wide Academies of Distinguished Teachers.
E-mail:kschug@uta.eduWebsite:www.chromatographyonline.com












LCGC Blog: Forensics Laboratories Underassess Uncertainty in Blood Alcohol Determinations
August 8th 2023The level of uncertainty provided by most forensic laboratories for reported blood alcohol results has been woefully underassessed. Not only is this bad science, but someone’s civil liberties may be at stake.


