The LCGC Blog: Chromatography Technical Tips - Robustness in HPLC Eluents
E-Separation Solutions
When developing a separation, some fundamental choices need to be made on how we might achieve the separation we require - primarily the mode of chromatography used and the way in which we intend to control and optimise the retention and separation (selectivity) of analyte components.
The ultimately robust HPLC mobile phase is 0.1% TFA (aq) / 0.1% TFA in Acetonitrile, right?
Maybe.
It would be great if we had arrived at a "global" HPLC mobile phase – but we haven’t, and here’s why, alongside some suggestions on achieving high quality separations with robust eluent systems.
When developing a separation, some fundamental choices need to be made on how we might achieve the separation we require – primarily the mode of chromatography used and the way in which we intend to control and optimize the retention and separation (selectivity) of analyte components.
In reversed phase mode, retention and separation will depend upon the physico-chemical properties of our analytes and knowing some of this information will help us to make more informed choices. This information will help us to make choices about the stationary phase bonded chemistry (C18, C8 etc,) which is needed – however this is outside the scope of this piece and I’m going to assume that a stationary phase has been chosen or that a range of phases will be screened to assess their suitability.
More hydrophobic analytes (Log P > 1) will require more organic modifier and less modifier is required for more polar, less hydrophobic analytes (Log P < 1). Selectivity is affected by the type of modifier used, which for many modern applications is either methanol or acetonitrile, both of which have different properties and interact differently with both our analytes and the stationary phase surface. Some simple separations may result in in using a simple binary mixture of water and one of these modifiers in either an isocratic or gradient programmed mode. But even in these simple situations we should be mindful of the pros and cons of the modifier and ways in which we can ensure the method is robust.
Different organic modifiers interact differently with analytes and the important properties which govern the selectivity of the common modifiers can be classified by their solvochromatic parameters. Dipole character π*, is a measure of the ability of the solvent to interact with a solute via dipolar and polarisation forces and will promote retention of polarisable analytes. Acidity α, is a measure of the ability of the solvent to act as a hydrogen bond donor towards basic (acceptor) solutes so will promote retention of bases. Basicity β, is a measure of the ability of the solvent to act as a hydrogen bond acceptor towards an acidic (donor solute), therefore, it will retain acidic analytes well. These characteristics, along with knowledge of the analyte chemistry, can be used to manipulate elution. In general, in a 50:50 mixture of solvent and water, acetonitrile will have a higher elution (eluotropic) strength than methanol.


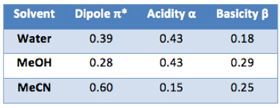
Table 1 – Solvophobic properties of various solvents regularly used in reversed phase HPLC

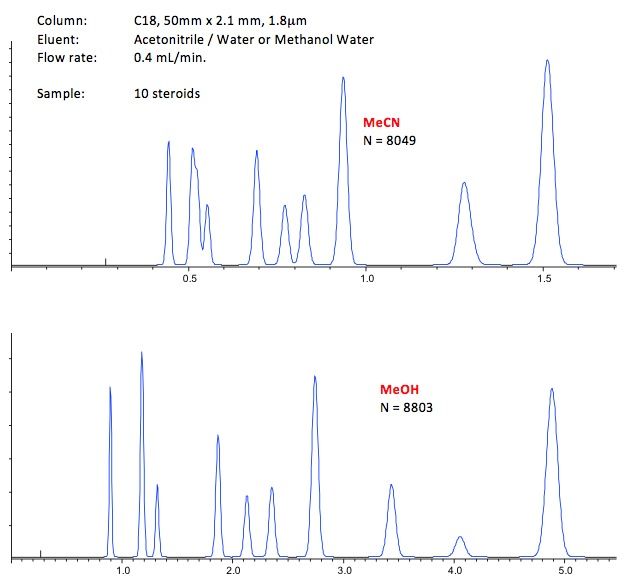
Figure 1 – Selectivity and retention differences observed in the separation of 10 steroid analytes with a range of hydrophobicity
Acetonitrile typically gives better efficiency at higher linear velocity (flow rate) as it is a lower viscosity solvent, which is good for higher throughput analysis. It also has a lower UV cut-off which can be beneficial when working at lower UV wavelengths.

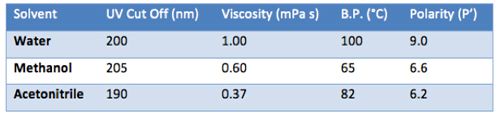
Table 2 – Physico-chemical properties of various solvents regularly used in reversed phase HPLC

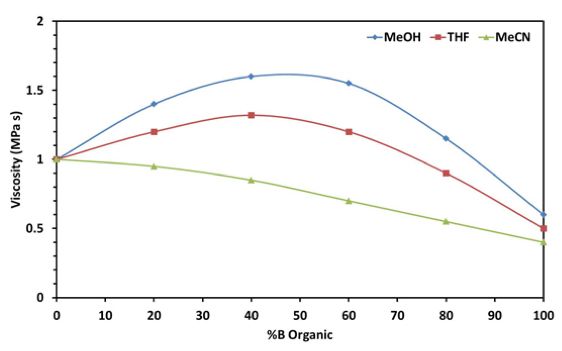
Figure 2 – Viscosity of various aqueous binary mixtures of solvents used for reversed phase HPLC
Certain combinations of methanol and water form high viscosity azeotropic mixtures which can affect use of certain solvent combinations as higher flow rates on instruments with lower back pressure limits.
When using pre-mixed mobile phases (both organic and aqueous portions combined in a single reservoir), one should always measure the relative amounts of each solvent required and then combine them rather than ‘topping off’ one solvent and making to volume with the other as exothermic events can mean that the ultimate aqueous / organic ration is incorrect. This is especially important with methanol water mixtures. Further, once mixed, one should guard against selective evaporation of the more volatile organic components which may lead to a gradual shift in retention time of analytes (typically to later elution times) over extended campaigns of analysis. Try not to filter pre-mixed mobile phases under vacuum, as again we risk loss of the more volatile organic component, which is an insidious and irreproducible problem.
If the analyte contains ionisable functional groups we may need to use pH to control the retention and selectivity of analytes. For acid species lower pH tends to result in longer retention and the opposite is true for basic analytes. The selectivity of the separation will depend upon the eluent pH and how close this value is pKa of the various analytes (the pH at which the ratio of ionised to non-ionised forms of the analyte is exactly 1:1). In this situation, two approaches are common. Adjust the pH to a high or low value to (2.2 or 10.0 are typical values in this regard), to ensure that all analytes are either fully ionised or fully non-ionised and the eluent pH is well away from the pKa of the various analytes OR carry out a series of experiments to find an eluent pH which gives good selectivity and resolution for analytes which may vary in their degree of ionization.
The former of these approaches guards against any problems with method repeatability, as if the eluent pH is close to to the pKa of any analyte, then only slight changes in eluent pH may lead to large changes in the retention time of that analyte relative to the others, the separation selectivity will change and resolution between closely eluting peaks may be lost. Of course, for analytes which are fully ionized, we need to ensure retention is still sufficient to avoid early elution, and retention factor values of greater than 2 should be aimed for to avoid the irreproducibility and poor resolution associated with elution in or near the void volume. This is typically achieved by lowering the amount of organic modifier or choosing a stationary phase which is capable of retaining more polar analytes which include polar end capped, polar embedded or those columns which have less hydrophobic bonded phases (phenyl, pentafluoroproply, cyanopropyl etc.).

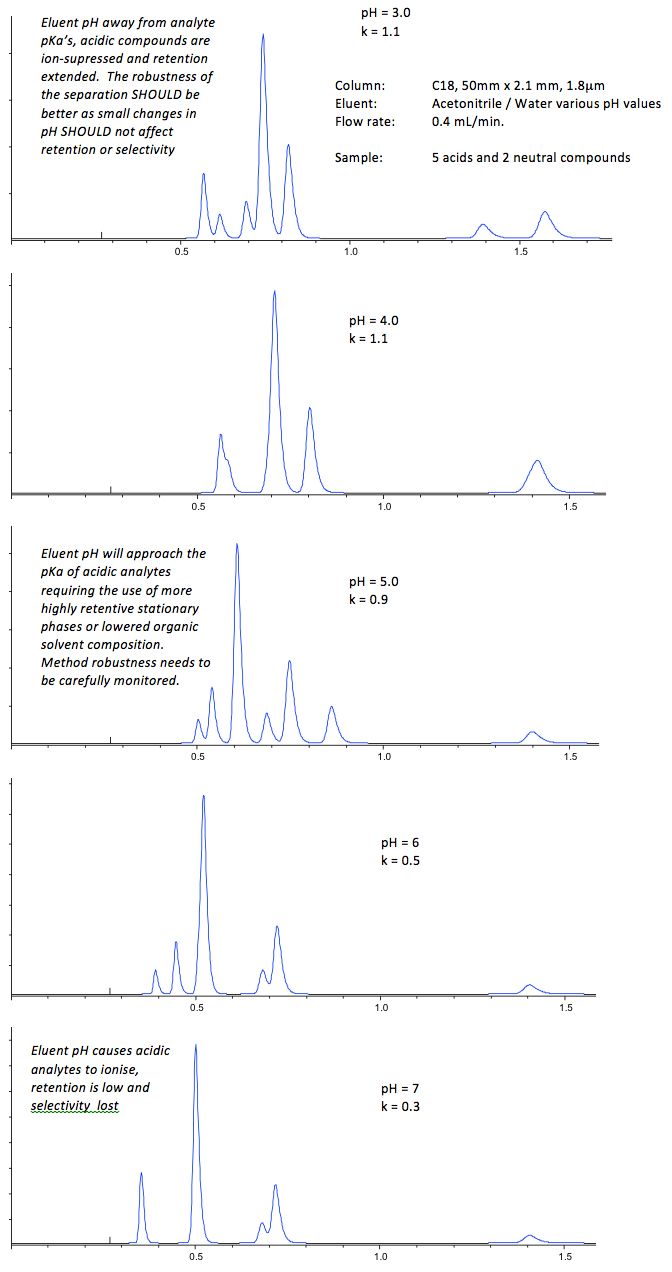
Figure 3 – Separations showing the various approaches to HPLC eluent optimization for ionizable compounds
The use of trifluoro acetic acid has become popular to adjust pH to around 2 (a 0.1% v/v solution of TFA results in a pH of around 2.1). TFA also has the benefit that it will form a neutral "ion pair" complex with ionised basic analytes (most basic analytes will be fully ionised at this pH) and therefore help to increase retention. When using TFA we should also be aware that it will be retained by the stationary phase whose surface will then be partially ionised, which has implications for column equilibration as well as the use of the column for other applications without being properly flushed. These multiple mechanisms mean that we still should not be blasé about our mobile phase preparation.
If using TFA to achieve a certain pH by adding it to water while monitoring using a pH meter, then the amount added (or more importantly the resulting hydrogen ion activity) may not the same as volumetrically adding TFA which would theoretically result in the required pH, as the initial pH of the water may be different. Depending upon the dominant mechanism by which TFA is controlling retention and selectivity, this may alter the separation between batches of eluent. Take care when using an acid (rather than a fully buffered system) that one always uses the same method to prepare the mobile phase and if changes in selectivity are noted – prepare the eluent volumetrically (0.1% v/v) rather than pH 'titration'. TFA will ion pair with basic species, so in +ve ion ESI LC-MS this acid species can cause drastic losses in signal intensity. The same problems are not true when using formic acid – however the gain in analyte retention may be appreciably lower, especially when analysing proteins and peptides. TFA typically also "lingers" within the MS ion source with ions at 69, 113, 127, 227 and 249 m/z in negative ion mode being indicative of TFA contamination.
Take care when using mobile phases containing volatile acidic or basic species as these may preferentially evaporate on standing, resulting in gradual changes in retention behavior. Remember to always cap eluent reservoirs with caps containing a small hole to prevent cavitation. Don't use laboratory film as plasticizers may leach into the eluent, which increases baseline noise and gives rise to artefact peaks, especially when using gradient elution and can result in increased spectral noise and interfering ions when using MS detection. Ions at 279 or 231, 522, 550 m/z in positive ion mode are indicative of plasticiser contamination.
Aqueous solvents will, over time, suffer from microbial growth even if they contain a buffer. This growth may contribute to system contamination and give rise to artefact peaks. For an aqueous buffered system, an expiry time of one week from preparation is generally accepted as being an appropriate and month from preparation for organic solvents.
It's better to use a fully buffered system to control the system pH, which is achieved by adding the conjugate salt into the eluent, alongside the acid or base. Ammonium formate with formic acid or ammonium acetate with trifluoroacetic acid can be used and one should be aware of the optimum buffering ranges of each buffer that is used (Table 3), which is ±1 pH unit either side of the buffer pKa value.


Table 3 – Physico-chemical properties of various buffers regularly used in reversed phase HPLC
If the buffer is incorrectly chosen, it will need to be added at much higher concentrations to be effective, which will lead to method robustness issues and changes in the selectivity of the separation that are more difficult to predict and manipulate.
In most situations, 5–15 mM buffer concentration will be sufficient for small molecule separations. Start with 25 mM of buffer and adjust lower if unpredictable selectivity changes are observed during method development and higher if a lack of robustness in retention time or poor peak shape is noted — otherwise it will remain unchanged! Note that at higher concentrations (especially above 60% acetonitrile), the inorganic buffers risk precipitation.
Just for the record, if one prepares a 10mM solution of ammonium acetate and then titrates the pH to, say, 2.9 with acetic acid (conc.), the absolute concentration of the aqueous solution will no longer be 10mM. This may become an issue if the selectivity of the separation is particularly susceptible to changes in buffer strength – but under normal circumstances, as long as we prepare the buffer in the same way every time, it shouldn't be a big problem.

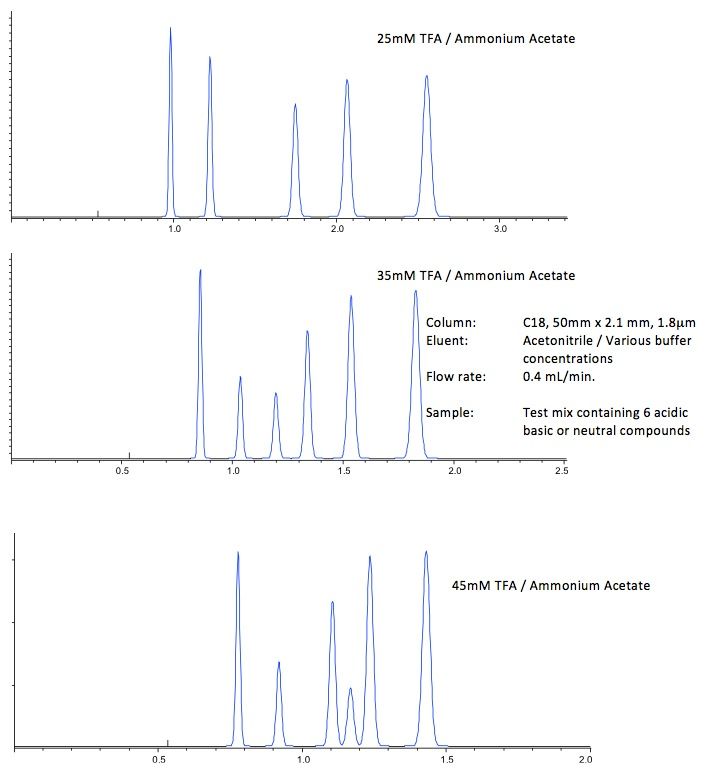
Figure 4 – Separations showing the change in retention and selectivity on changing the ionic strength of the buffer
If ion-pairing or buffer surface interactions are highly important and you suspect this may be contributing to robustness issues, one should prepare separate 10mM aqueous and 10mM organic solutions so that, when combined in any ratio, the absolute ionic strength remains constant. This is often seen in gradient HPLC when 0.1% (aq) and 0.1% v/v TFA in acetonitrile are combined to maintain ionic strength across the gradient – as suggested in the title to this piece.
We also, actually don't know the actual pH of the eluent or indeed the pKa of any of our analytes once we mix the aqueous and organic portion of our eluent, and depending on the buffer and the organic used, these may go either up or down in real terms. Hence the value in consistency of eluent preparation, both within and between laboratories, were small variations in sample and eluent preparation can often lead to insidious problems with method transfer.
An improperly washed pH meter probe can result in relative large amounts of carryover of acidic or basic species – resulting in changes to the final hydrogen or hydroxide ion activity within the eluent from batch to batch – potentially resulting in changes to the retention and selectivity. Also – just a reminder here to ensure that the pH meter is properly calibrated using pH buffers which straddle the expected pH of the eluent you are preparing.
Finally, when working out the cause of problems with retention time and trying to decide if hardware or mobile phase changes are to blame, try overlaying the chromatograms and comparing the injection solvent disturbance peaks alongside the analyte peaks of interest. If the void peaks overlay well but the analyte peaks do not, then the mobile phase composition must have changed in some way.

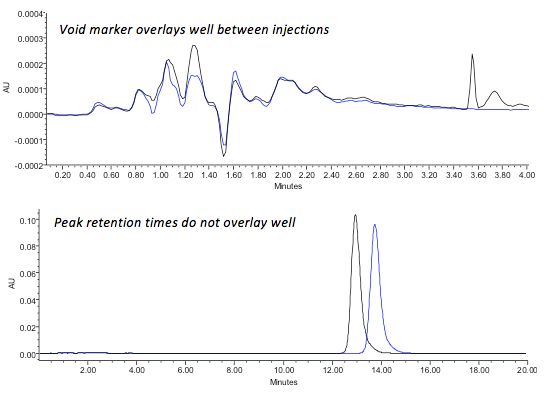
Figure 5 – Comparison of void marker and analyte retentiondrift to identify potential changes caused by eluent composition in reversed phase HPLC separations
For more information – contact either
Bev ([email protected]) or Colin ([email protected]).
For more tutorials on LC, GC, or MS, or to try a free LC or GC troubleshooting tool, please visit www.chromacademy.com



.")


The LCGC Blog: Historical (Analytical) Chemistry Landmarks
November 1st 2024The American Chemical Society’s National Historic Chemical Landmarks program highlights sites and people that are important to the field of chemistry. How are analytical chemistry and separation science recognized within this program?



