Interpretation of Isotope Peaks in Small Molecule LC?MS
Biologists entering liquid chromatography?mass spectrometry (LC?MS) from a background of general LC rarely use their mass spectrometer to its full potential. Isotope peaks offer huge possibilities both in semi-quantitative interpretation of structure and in quantitative labelling studies. This article examines charge state, "easy" isotopes, such as chlorine, the slightly harder problem of sulphur compounds, and finally looks at a method for improved measurement of heavy labels in a metabolic study.
Biologists entering liquid chromatography–mass spectrometry (LC–MS) from a background of general LC rarely use their mass spectrometer to its full potential. Isotope peaks offer huge possibilities both in semi-quantitative interpretation of structure and in quantitative labelling studies. This article examines charge state, "easy" isotopes, such as chlorine, the slightly harder problem of sulphur compounds, and finally looks at a method for improved measurement of heavy labels in a metabolic study.
Introduction
Mass spectrometry (MS) is becoming commonplace in biological laboratories. In many ways, liquid chromatography–mass spectrometry (LC–MS) presents few problems to the biologist with general experience of LC. The mass spectrometer is merely a new detector, and conveniently, it operates in units that are already familiar, those of molecular mass [this contrasts starkly with the ppm axis of nuclear magnetic resonance spectroscopy (NMR)]. However, MS offers many possibilities beyond simple detection. This article looks at one such possibility, the study of isotope patterns. Heavy isotopes, both as labels and at their natural abundance, can be useful even without recourse to sophisticated instruments designed for their accurate measurement.
Their interpretation is not immediately obvious to those without a background in analytical chemistry, and their discussion in textbooks is often scanty, probably limited to charge-state measurement, and the use of isotopically-labelled internal standards. These applications are merely the tip of an iceberg. Using a range of examples, I aim to show how a proper consideration of isotope patterns can contribute greatly to the understanding of small-molecule (i.e., non-proteomic) biological LC–MS data. I place special emphasis on ion trap LC–MS, as ion traps are a popular, relatively inexpensive entry into the world of MS–MS, but require a little care if their full potential is to be realized. Nevertheless, the techniques described in this article are applicable to a wide range of LC–MS instruments.
Mass as Measured by a Mass Spectrometer
Mass spectrometers measure the mass per unit charge (m/z) of an ion. Using any of the common LC–MS ion sources, small molecules will often have a charge (z) equal to one, and the ion will be created by some simple process such as the addition of H+ , accompanied by a very predictable change of mass from molecule to pseudomolecular ion. In such instances, the mass spectrometer effectively tells us the mass of the molecule. Although the mass may well be quoted in amu (atomic mass units) or Thompsons, we can treat it as a mass in Daltons and match it up to relative molecular masses in chemical catalogues, and the match will be reasonably accurate.
However, a glance at the spectrum will show smaller peaks slightly heavier than the pseudomolecular ion. These are the isotope peaks and their existence is the reason why there is only an approximate match between the mass spectrometer and the chemical catalogue. The mass spectrometer measures the mass of each ion it encounters; if an ion contains a heavy isotope instead of the normal one (13C instead of 12C), then it will be recorded separately at the heavier mass. Since many elements have fairly common heavy isotopes (carbon is about 1% heavy), the isotope peaks are not insignificant. The chemical catalogue, however, lists molecular masses designed to allow the weighing out of known molar quantities so it contains the mass of one mole of the natural mix of isotopomers (molecules that are identical in every way except that they contain different arrangements of heavy isotopes). Common elements with a large difference between exact mass and average molar mass include chlorine, with a relative atomic mass of approximately 35.5. This arises because chlorine is actually approximately ¾ 35Cl and ¼ 37Cl, which the mass spectrometer measures as two peaks, differing in mass by 2 amu, and present at relative intensities of 3:1. Tables of true masses and intensities are available in data handbooks, textbooks,1 and software.
Having realized why isotope peaks exist, many biological users promptly ignore them. This is unfortunate; heavy isotopes are extraordinarily useful, both at their natural abundance and in specifically enriched, labelled compounds.
The most traditional use of isotopes is in internal standards. Since the mass spectrometer measures mass, it can be used as a simple mass-selective detector. It is hard to surpass the sensitivity of a simple single quadrupole mass spectrometer in single ion monitoring mode (SIM), where instead of scanning all masses, the instrument is set to measure continuously a handful of target masses. A compound identical to the analyte of interest, but labelled with heavy isotopes, can be detected in SIM independently of the native analyte, but will have identical chemical properties, with the same efficiencies of extraction and ionization. It is the perfect internal standard. I will not discuss this here as internal standards will be familiar to readers from most LC backgrounds, and are covered in any good textbook.
Measuring Charge State
This is another well-documented, classic use for isotope peaks, but worthy of a closer look, especially in ion traps. Almost all analytes will contain carbon, and carbon has a convenient +1 isotope (i.e., a heavy isotope weighing 1 Da more than the main peak). Therefore, almost all analytes will have isotope peaks spaced regularly at intervals of 1 Da (according to whether 1, 2, 3 etc. of the carbons are 13C). But mass spectrometers measure mass per charge (m/z), so if the charge state (z) is two, the isotope peaks will occur at intervals of +½, +2/2, +3/2 etc., intervals of 1/2. If the ion had a charge of 3, the peaks would appear at intervals of 1/3. The spacing of the isotope peaks is, therefore, used to measure the charge state of the ion, and the charge state is then used to find the actual mass of the ion. For example, an analyte that appears at m/z = 400 amu with a charge state (z) of 2 has a mass (m) of 400 × 2, or 800 Da.
In practice things can be less clear. Figure 1(a) shows a spectrum from an organometallic compound analysed in a Thermo Finnigan DecaXPplus ion trap in its "zoom scan" high resolution mode. This is not an accurate mass spectrometer, but nevertheless resolves to 3 or 4 peaks per amu. This spectrum is not clean; the peaks are not fully resolved and at a glance it is not clear what charge state this molecule has. Counting peaks between 825 amu and 826 amu, the region where the spectrum is well resolved but the peaks are big, there are 4 peaks (suggesting a charge state of 4). But at higher masses there are clearly 3 peaks per amu. At low mass, the peaks become blurred and resolution is insufficient to count. This is typical of ion traps, in which there is a pay-off between signal strength and resolution. When the trap contains too many ions, they interact; each individual ion no longer experiences exactly the electric field applied to the trap, but is also influenced by the electric fields of its neighbouring ions. This reduces resolution.



Figure 1
During scanning, ions leave the trap in order of increasing mass. The lightest ions leave first, at a time when the trap is fully populated, so they are most subject to loss of resolution and distortion of peak shape. By the time the heavy ions come to leave the trap, their light neighbours, which would have influenced them, are already gone. They, therefore, scan with better resolution. For this reason, the high mass end of the isotope cluster is the best area to count. Of course a better alternative may be to reduce the target ion count in the trap, which will reduce signal (worsen signal-to-noise ratio) but improve resolution. This is, in fact, the difference between the spectra in Figure 1(b) and 1(c). Those who work only with time of flight (ToF) or quadrupole systems will not encounter this problem.
In the previous paragraph I referred to the high resolution mode of the DecaXPplus ion trap. Resolution and mass accuracy are beyond the scope of this article but must be understood. Resolution is the ability of an instrument to separate two ions of nearly identical mass and show them as two, separate, narrow peaks. Even without accuracy, good resolution is useful for counting charge state and examining individual ions. An accurate mass instrument almost certainly has high resolution, but in addition it measures the position of a peak (i.e., the mass of an ion) with great precision. If properly calibrated, the mass will not only be precise, but accurate (i.e., correct), the usefulness of which I will return to later.
Charge state measurement is also relevant in MS–MS analyses. Manufacturers of MS–MS instruments rightly promote their ability to isolate a parent ion uniquely prior to fragmentation. However, by reducing the resolution with which the parent ion is trapped (increasing the isolation width), it is quite possible to trap an entire isotope cluster. This is useful in measuring mass losses when the precursor ion is multiply charged. Fragments with a lower m/z than their multiply-charged (z > 1) precursor ion may have lost charge as well as mass. A doubly-charged precursor ion of 400 amu (m = 800 Da) can give rise to a fragment of 300 amu, which could have a charge of 1 (and m = 300 Da) or 2 (and m = 600 Da). Hence the true mass loss could be 500 Da or 200 Da, two possibilities that can be distinguished by looking at the isotope spacing of the fragmention.
Counting Atoms of "Easy" Elements
Not all elements have good abundances of heavy isotopes. Carbon has only about 1% of 13C. Of the common biological elements, oxygen has a very small +2 isotope peak, and sulphur is the only respectable element with a +2 peak of about 4%. I will look at sulphur in more detail later. Easy elements include many transition metals and some halogens. These both have relevance in biological analysis.
Fluorine has no isotope peaks but chlorine is about 25% 37Cl, while bromine contains approximately equal amounts of 79Br and 81Br. Halogen peaks are sufficiently clear that they can be measured in a simple mass spectrometer with unit mass resolution, and analysed without complicated software. It is often possible to determine by eye from the pattern how many halogen atoms are involved. Figures 2(a) and 2(b) show spectra from tribromophenol, before and after treatment with a peroxidase from a species of duckweed with an ability to detoxify trihalophenols in its surroundings.2 Note that the parent tribromophenol shows four peaks [Figure 2(a)] corresponding to all combinations from no heavy bromine to three heavy bromine atoms. Figure 2(b) shows the singly-debrominated version, with only three peaks. These spectra were derived from LC–MS runs using an Agilent single quadrupole mass detector, illustrating that the use of isotope patterns is not restricted to instruments sold specifically for high-resolution scanning or accurate isotope analysis.

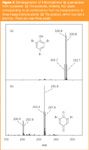
Figure 2
Bromine is very easy to interpret with simple mathematics because the two isotopes are roughly equally common. It brings to mind school probability questions about a selection of three coloured marbles from a bag containing equal numbers of blue and red: in this instance there are three ways to arrange a molecule with either one or two heavy atoms, and only one way to arrange the molecules with no or three heavy atoms. Therefore, the middle peaks are three times the intensity of the outer peaks.
Similar calculations can be performed for any isotope and tools for calculating isotope patterns are now common in mass spectroscopy packages. Figure 3(a) shows a nickel-containing molecule synthesized as a model of an enzyme active site.3 This is a large, complex molecule, and the calculation of its predicted isotope pattern requires a software tool; we used Thermo Finnigan's isotope viewer, which converts single masses into Gaussian curves to allow easier comparison with real experimental data. The match between expected [Figure 3(b)] and measured data [Figure 3(a)] is good. However, without knowing the original structure it is still possible to guess the number of nickel atoms involved. The isotope peaks at even mass increments above the base peak are dominated by the nickel [shown bold in Figure 3(b)], while the peaks in between are mostly the result of 13C. Comparison of the bold, +2 peaks with the isotope patterns of one and two nickels (inset) shows that this molecule must contain two.


Figure 3
Isotope pattern recognition, appropriately automated, thus has a part to play in the determination of empirical formula. Accurate mass is conventionally used to calculate empirical formulae. This works because no atom apart from 12C actually weighs an exact integer, and, therefore, many chemical groups that weigh approximately the same can actually be distinguished if the mass spectrometer is accurate enough. For instance, an H2PO3 group has an exact mass of 80.9742 Da while HSO3 has a mass of 80.9646 Da. These masses differ by approximately 0.01 Da while typical quadrupole and ion trap systems offer precision no better than 0.1 amu. The accuracy of accurate mass instruments, which are usually time of flight (ToF) or FT–MS instruments, is often quoted in parts per million (ppm). This value depends on the mass of the ion being measured, and, therefore, ppm must be specified at a particular mass. ToF instruments can achieve 5 ppm for masses above 600 amu, and FT–MS is more accurate still. But even at 5 ppm accuracy, there will still be a list of likely formulae rather than a single result. The list can be pruned using chemistry (the nitrogen rule) and common sense. In addition, systems such as Bruker's MicrOTOF, with "True Isotopic Pattern" fit use the isotope pattern to rank the results in order of likelihood. The isotope pattern is, therefore, a piece of information that can be used in conjunction with accurate mass, to improve certainty of results.4
Counting Sulphur Atoms
While it is easy to see the large isotope peaks of halogens and metals, in our experience it is harder to be sure of intermediate elements such as sulphur. The 4% +2 peak can easily be obscured by poor resolution or noisy background signal, especially if it is added to the non-sulphur +2 signal of a molecule that is very large (with a high probability of containing two 13C atoms), or contains a lot of oxygen (which has its own small +2 peak). A small change in a small peak, subject to large errors, is hard to see. Fortunately, using MS–MS this can often be converted to an almost qualitative change in a very large peak. The method is to select for analysis the +2 isotope peak. This contains one heavy sulphur. Then we search for a fragment that has lost a sulphur. If there is only one sulphur present, then the sulphur that has been lost must have been the heavy one, and the fragment will not be heavy, despite its heavy parentage. If, however, there are two sulphurs present, then there is an equal chance of losing a light or heavy sulphur. Therefore, there will be two fragment peaks of roughly equal intensity, one heavy and one normal. In this way, a small change in a 4% abundance of +2 peak is converted to a presence/absence change in a very clean MS2 spectrum. Of course when more than two sulphurs are present, the ratio of light and heavy fragment will reflect the number of sulphurs. This useful method can be found in textbooks, but rarely treated systematically.
Figure 4(a) shows the MS2 spectrum of glucosinalbin, which belongs to the glucosinolates, a group of chemicals responsible for flavours in Brassicaceae , and implicated in defence against pests, as well as human nutrition. All glucosinolates share a common structure with two sulphurs and, therefore, we wanted a reliable way to check for this. Figure 4(b) shows the corresponding MS2 spectrum of the +2 parent at 426 amu, which was isolated with no detectable contamination by 424 amu. Note that the original fragment of 259 amu is now accompanied by nearly equal amounts of 261 amu. Even without knowing the identity of the fragment, it is clear that the fragment included at least one sulphur, from a parent that contained at least two. Further, the near 1:1 ratio of heavy and light fragments suggests that the fragment contained nearly half the total sulphur. Helpfully, there is also an original fragment of 97 amu, showing a very similar effect [Figure 4(b) inset]. We can almost certainly assign this to HSO4- , a single sulphur. Therefore, the remainder of the molecule also contained a single sulphur, two in total.


Figure 4
The alert reader will have noticed that the peak of 97 amu in Figure 4(b) (inset) is actually somewhat larger than 99 amu. This is a real effect and can be calculated properly. Some of the heavy parent ion at 426 amu is heavy not because it contains 34S, but because it contains 18O or two atoms of 13C. Since the HSO4- fragment contains less than half the total oxygen, and none of the total carbon, these alternative heavy precursor ions will mostly lead to light fragment ions, causing a slight bias. Table 1 shows the approximate amounts of +2 isotope peak caused by 34S, 13C and 18O, and their fate on fragmentation. The relative abundance of +2 peak caused by two 13C atoms is calculated as follows: the relative abundance of the +1 species is given by multiplying the relative abundance of 13C atoms by the number of atoms that might be labelled (1.1% × 14). Then we repeat the calculation to find the abundance of the +2 peak, where there are 13 atoms remaining that could now be made heavy. The abundance is now (1.1% × 14) × (1.1% × 13). But we have counted each isotopomer twice, because we could have added heavy carbon at the 7th position (for example) and then the 9th, or at the 9th and then the 7th, and the two products would be the same. Therefore, we divide the final abundance by two. (Details of isotope peak calculation can be found at our website: http://www.jic.ac.uk/Services/Metabolomics/interp/isocalc/isocalc.htm). The calculated amount of 99 amu heavy fragment is about 80% of the amount of 97 amu light fragment, a result which agrees well with the experimental data. For those who wish to avoid the arithmetic, had there been three sulphurs, the ratio between the two peaks would have been 2:1, and the 99 amu peak would have been half the size of the 97 amu peak. It is clearly larger than this, so qualitatively there were only two sulphurs.


Table 1
This method of sulphur-counting is highly reliable and our laboratory has successfully used it on analytes as diverse as plant-derived isothiocyanates and synthetic sulphur-containing organometallics.
This glucosinolate molecule offers one more example to see how sulphur isotope peaks can be used. MS3 analysis of the 259 amu fragment showed that it fragmented in a way very reminiscent of glucose phosphates (Figure 5(a) and inset). We, therefore, concluded that there had been an intramolecular rearrangement to transfer the glucose to the sulphate group, which has approximately the same mass as a phosphate group. This could be confirmed by trapping the 261 putative heavy glucose-34SO4 fragment [Figure 5(b)] and noting that in MS3 it produces H34SO4- at 99 amu, and very little H32SO4- at 97 amu (the latter being ascribed to 261 amu precursor ion that was heavy for reasons other than 34S, as above). A genuine heavy glucose phosphate isotope peak would owe its heaviness mostly to 13C and 18O (of which less than half is present in the phosphate group), and would, therefore, produce more light phosphate than heavy phosphate. In fact, this conclusion also allows us to identify the 275 amu MS2 fragment of glucosinalbin as an analogous glucose-S-SO3- rearrangement, in which no sulphur has been lost, agreeing with the observation that the +2 parent always produces a +2 fragment at 277 amu, never a light fragment at 275 amu [Figure 4(b)].


Figure 5
I hope that this example has shown how a careful and logical examination of the fragmentation behaviour of a +2 parent can give a lot of information about the sulphurs present. Note that this is complementary to such techniques as proton NMR, which may yield limited information about chemical groups devoid of hydrogen (isothiocyanates etc.).
Working with Stable Isotopes
All the examples given look only at isotopes present at their natural abundance. Labelled compounds, enriched in some isotope, have a very long history in metabolic studies. Traditionally radioisotopes are used for high sensitivity work and stable isotopes such as 13 C are common in NMR-based studies where the position of the label can be used to deduce how the analyte has been metabolized. Unfortunately, it is usually very tedious to find the position of radiolabel in a molecule, and NMR often lacks sensitivity. The use of stable isotopes in LC– or GC–MS occupies a middle ground, combining reasonable sensitivity and limited ability to differentiate between isotopomers.
Again, however, accurate measurement of a labelled peak can be difficult when the background is noisy and there is a very large peak of unlabelled material only 1 amu below the labelled mass of interest.
Here I present a method that we have used to analyse 15 N-labelled amino acids in seeds of oilseed rape using LC–MS. Our approach was to tag the amino acids using a commercial tag (Waters AccQ tag). This permits good separation on a C18 column without resort to ion pairing reagents (which in our experience can be hard to remove from a spray chamber, interfering with future work to an undesirable extent). It also permits absolute measurement of amino acids by absorbance or fluorescence of the tag. A naive approach would be to quantify the labelled amino acid by measuring the intensity of the +1 peak. This does not work, because even unlabelled amino acids have a considerable +1 peak, exacerbated by the large amount of carbon in the tag. We would, therefore, be looking for small increases in a large and noisy number.
Our alternative strategy was to trap, specifically, the +1 peak, and fragment it. This already has a superficial benefit of removing a large part of the carbon responsible for the unwanted +1 signal. The majority of tagged amino acids lose the amino acid as a neutral leaving group, and the detected fragment is then tag. Detection of tag rather than amino acid does not matter: a light tag-fragment, from a heavy tagged amino acid, must indicate that the parent ion contained a heavy amino acid. In fact, this method benefits enormously from the tag, because we can use it as an internal standard. We can calculate very accurately the natural incidence of heavy tag relative to light tag, allowing us to calculate the percentage enrichment of isotope in the amino acid. The arithmetic is as follows:


Figure 6
The instrument will usually measure target metabolite relative to an internal standard. In this instance it is measuring


Where A and T are amino acid and tag, and the subscript indicates heavy or light. AhTl is, thus, the intensity of heavy amino acid linked to light tag. Normally, the measured value would be converted into a concentration using an external standard calibration curve of known amino acid concentrations and known internal standard. In this instance we used an external calibration "curve" of a single point from a mixed amino acid standard, and set the concentration to one "unit". The instrument software, therefore, calculated a signal:

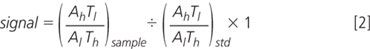
The amount of a heavy labelled compound is equal to the total amount of the compound multiplied by the probability of it being heavy.

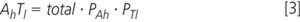
Substituted into the equation for "signal", cancelling the total amount of compound:

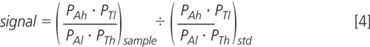
The probability of tag being labelled is constant and, therefore, cancels between standard and sample:

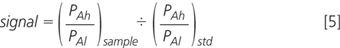
But PAh/PAl is actually the relative intensity of the isotope peak, which can be calculated from the numbers of atoms of each element present, and the relative abundances of the heavy isotopes (available in any good text book). Working in percentages

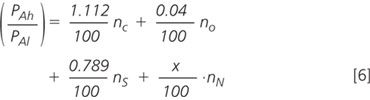
where x is the relative abundance of heavy nitrogen (which we are trying to measure), and the subscripts to n indicate the element. The non-nitrogen heaviness is a constant (we have not labelled anything except the nitrogen; this constant will become k). Furthermore, in the standard, which is not labelled, we know that x is 0.37%. Therefore, the measured signal is actually equivalent to

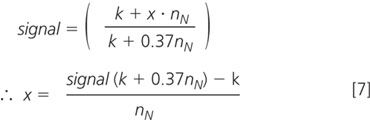
Since we can calculate k and nN is known for each amino acid, we can use this equation to calculate the relative enrichment of nitrogen in an amino acid as a percentage relative to the light form (i.e., the number of heavy atoms present for every 100 light atoms).


We may prefer to calculate the percentage of all nitrogen that is heavy (i.e., the number of heavy nitrogen atoms per 100 atoms in total, or, working in fractions, the number of heavy nitrogen atoms per 1 atom in total). For the fraction version rather than the percentage:

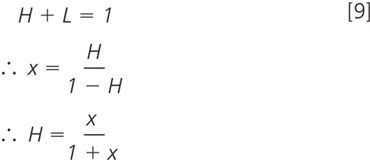
We have used this method with the Thermo Finnigan LCQ DecaXPplus ion trap to generate the sample data shown in Figure 6; similar methods would work for derivatized analytes other than amino acids, and would work in other LC–MS–MS systems. In fact ion trap users will have to be more careful than those with Q–ToF or triple quadrupole systems. It is obviously necessary to start with the +1 isotope peak isolated free of contamination by the light peak. Ion traps tend to trap with a lower resolution than they scan. The reason is that the scanning mechanism usually combines any useful method (ramping trap AC combined with resonant ejection) to empty the trap completely, in order of mass. Trapping requires the ejection of ions both smaller and larger than the target while retaining the target itself. This limits the options for ejection (e.g., resonant ejection alone) and thus the quality of resolution.
Conclusions
It would be impossible to cover all possible uses of heavy isotope peaks in a short article; in fact I have made no attempt to address the myriad of applications in proteomics. However, the applications above illustrate the value of isotope peaks in the study of halogens, sulphur and metals, and especially how isotope analysis can be combined with MSn in structural elucidation. The method presented for amino acid analysis shows how existing LC methods can be modified with very little extra interpretation to yield MS-methods of great value in labelling studies.
Acknowledgments
Organometallics (Figures 1 and 3) were synthesized and analysed by Sam Duff and Dave Evans. I also thank Chris Pickett and his group for introducing us to transition metal isotopes. Duckweed dehalogenation (Figure 2) was the work of Marcel Jansen and Roger Thorneley and its interpretation benefited greatly from the help of Fred Mellon (Institute of Food Research, Norwich). Analysis of glucosinolates was in collaboration with Stan Kopriva. Amino acid 15 N incubation and derivatization was performed by Olivia Lepri in the group of Steve Rawsthorne. I thank all the above collaborators in the John Innes Centre for their support and permission to use their data, and also Trevor Wang, Alison Smith and Baldeep Kular (John Innes), and Mark Bennett (Imperial College) for helpful comments on the manuscript. Our laboratory is funded by the UK Biotechnology and Biological Sciences Research Council (BBSRC), to whom we are grateful.
Lionel Hill trained as a biochemist, obtaining his PhD from the University of East Anglia/John Innes Centre. After some years working in plant primary metabolism and the biochemistry of potatoes and oilseed rape he turned to an analytical career, and now specializes in LC–MS for the John Innes Centre metabolite service (Norwich, UK).
References
1. E. de Hoffmann and V. Stroobant, "Mass Spectrometry, Principles and Applications" 2nd Ed., Wiley (2002).
2. M.A.K Jansen, L.M. Hill and R.N.F. Thorneley, Plant Cell and Environment, 27, 603–613 (2004).
3. S.E. Duff et al., Inorganic Chemistry Communications, 8, 170–173 (2005).
4. S. Ojanperä and I Ojanperä, LCGC Eur., 18, 607–614 (2005).



















