Immunoaffinity Solid-Phase Extraction with HPLC-FLD Detection for the Determination of Aflatoxins B2, B1, G2, and G1 in Ground Hazelnut
Special Issues
Using highly specific immunoaffinity solid-phase extraction to selectively extract and enrich aflatoxins prior to HPLC-FLD analysis, without derivatization, low LOQ values-far below the European regulations-can be achieved even in highly complex and fatty matrices.
Aflatoxins, a group of mycotoxins, are highly toxic substances and very harmful to human health. Consequently, the European Commission has set very low maximum levels for aflatoxins in various foodstuffs. Aflatoxin B1 is considered to be extremely toxic to human health, but also the aflatoxins B2, G2, G1, and the milk-derived derivatives M1 and M2 are in focus and demand rigorous analytical control. Trace-level quantification of these toxins requires very sensitive and reliable analytical methods. This work describes the determination of four aflatoxins in ground hazelnut by immunoaffinity solid-phase extraction (SPE)–based sample preparation and subsequent HPLC measurements with fluorescence detection without pre- or post-column derivatization. The use of a selective immunoaffinity-SPE and fluorescence detection enables very low detection limits to fully meet the requirements. The separation was achieved on a C18 column within 4 min under isocratic condition. Good linearity and recovery rates qualify the method as a fast, reliable and selective solution for the quantitative determination of aflatoxins in ground hazelnut.



(PHOTO CREDIT: PHOTOCREW/STOCK.ADOBE.COM)
Mycotoxins are naturally occurring fungal toxins that were first found in the fungus aspergillus flavus. The aflatoxins (AFs) are one prominent subgroup, of which 20 naturally occurring forms are known. AF–B2, AF–B1, AF–G2, and AF–B1, as well as the milk-derived variants AF–M1 and AF–M2, are the major compounds out of this group. Nuts, grains, herbs, and spices are common sources of aflatoxin contamination, while the M derivatives are found in dairy products. In particular, AF–B1 is considered to be a very potent carcinogen, which mainly affects the liver, and may therefore cause liver cancer, as well as chronic hepatitis, jaundice, cirrhosis, and impaired nutrient metabolism (1). Due to their high thermal stability, they are not destroyed during the production process or cooking procedures (2).
The European Commission has set various maximum levels for aflatoxins in several foods under consideration of their consumption and use. The maximum level for aflatoxin B1 ranges from 2 to 12 µg/kg for foods used for direct consumption or as ingredients, with the exception of baby food products with a maximum level of 0.10 µg/kg. Secondly, the sum of AF–B2, AF–B1, AF–G2, and AF–G1 must not exceed 4 to 15 µg/kg (3). As a result, a sensitive and accurate analytical method is required to control the levels of these toxins in various foodstuffs.
Whereas thin-layer-chromatography (TLC) was previously used for the determination of B and G aflatoxins, nowadays high performance liquid chromatography (HPLC) coupled to fluorescence detection (FLD) with and without derivatization, as well as mass spectrometry (MS), is commonly applied (4). While MS provides additional peak identification potential, it is a relatively costly technique that requires a certain level of qualification of the analyst, and is therefore not available in all food analysis laboratories in emerging countries. On the other hand, derivatization techniques require additional instrumental effort as well, and may also negatively affect method robustness and transferability.
A powerful alternative to increase the method specificity and its limit of quantitation (LOQ) is the use of highly specific immunoaffinity solid-phase extraction (SPE) to selectively extract and enrich the aflatoxins prior to the HPLC–FLD analysis (5). With this sample preparation method, low LOQ values with direct fluorescence detection can be achieved, even in highly complex and fatty matrices.
This article describes the determination and quantification of AF–B2, AF–B1, AF–G2, and AF–G1 in ground hazelnut with immunoaffinity SPE purification followed by HPLC–FLD analysis without derivatization. The applied technique allows sufficient trace level detection for this matrix far below the European Commission regulations.
Experimental
Chemicals and equipment
Methanol (LC–MS grade), acetonitrile (LC–MS grade), hexane (HPLC grade), phosphate buffered saline (PBS) buffer pH 7.2, acetic acid (LC–MS grade), and sodium chloride were obtained from Fisher Scientific. The aflatoxin mixture consisting of B2, B1, G2, and G1 was purchased from Sigma Aldrich. AflaClean Select Immunoaffinity SPE cartridges were purchased from LCTech GmbH.
Preparation of Standards
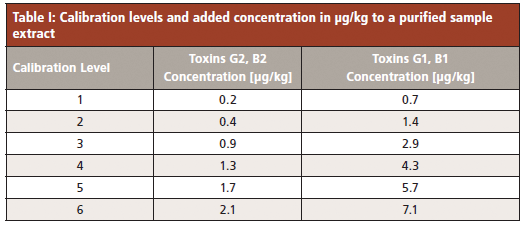
Quantification was performed by standard addition calibration to correct matrix influences, due to the absence of a hazelnut sample that was free of any aflatoxin content. The aflatoxin stock solution (stock solution 1), containing AF–B2 and AF–G2 with a concentration of 857 µg/kg, and AF–B1 and AF–G1 with a concentration of 2856 µg/kg, was used to prepare the calibration standards by diluting stock solution 1 with 1% acetic acid in the ratio 1:100 (stock solution 2).
Stock solution 2 was added to a purified sample extract after immunoaffinity SPE (see "Preparation of Samples," below) to obtain different calibration levels, as listed in Table I. The recovery experiment was carried out by adding 113 µL of the aflatoxin stock solution 1 to 20 g of the ground hazelnut sample prior to sample preparation. The resulting concentration of the recovery sample is 1.7 µg/kg for G2 and B2, and 5.65 µg/kg for G1 and B1, when 100 % recovery is assumed.

Preparation of Samples
Spiked and nonspiked samples were prepared in triplicates. Then, 20 g of ground hazelnut sample and 2 g sodium chloride were weighed into a 200 mL bottle and 100 mL 80% methanol and 50 mL hexane were added. The solution was stirred for 15 min, and then filtered through a folded filter. Thereafter, 14 mL of the lower liquid phase was mixed with 86 mL PBS buffer pH 7.2 and 50 mL was loaded onto the immunoaffinity SPE cartridge. Afterwards, the cartridge was washed with 10 mL of water. Elution was carried out with 2 mL methanol, with the first 1 mL applied to the cartridge and allowed to react for 5 min. The extract was diluted 1:2 with 1% acetic acid before it was filtered through a 0.2 µm regenerated cellulose membrane in the HPLC vial for injection.
Instrumentation
The Thermo Scientific Vanquish Flex UHPLC system consisted of a quaternary pump, split sampler, column compartment and fluorescence detector, equipped with a standard bio flow cell with 8 µL volume. Chromatographic separation was performed on a 100 × 3 mm, 3 µm Thermo Scientific Acclaim C18 column, using a 4 min isocratic method of 50% water, 30% methanol and 20% acetonitrile with a flow rate of 0.5 mL/min. The injection volume was set to 20 µL. The active preheater and the column temperature were held at 30 °C constantly. The FLD excitation and emission wavelength were set to 365 nm and 450 nm, respectively, while the lamp was operated in high power mode with a sensitivity of 8. The data acquisition and processing was performed with Thermo Scientific Chromeleon 7.2.8 chromatography data system (CDS) software.
Results and Discussion
During the method development process, various aqueous and organic mobile-phase combinations were tested in order to achieve sufficient baseline separation of the four toxins AF–B2, AF–B1, AF–G2 and AF–G1 on the C18 column. The best result could be obtained with 50/30/20 water/methanol/acetonitrile (v/v/v) mixture with a flow rate set to 0.5 mL/min. At this point, no immunoaffinity SPE clean-up prior to HPLC-FLD analysis was performed. Figure 1 shows the separation of the aflatoxin mixture with excitation at 365 nm and emission at 450 nm. Some peaks were eluted before the first target aflatoxin G2. These unknown peaks are impurities in the standard solution that have not been further identified. All aflatoxin analytes are baseline separated within 4 min.


Figure 1: FLD chromatogram of AF–G2, AF–G1, AF–B2, and AF-B1 at concentrations of 0.9 µg/kg for AF-G2 and AF-B2, and 2.9 µg/kg for AF-G1 and AF–B1.
Calibration was performed with the standard addition method. The original sample, which was found to already contain all analytes, was set to zero amount, which results in a negative x-axis intercept. In this way, the calculated amount of the analytes corresponds to the absolute amount of the negative x-intercept. Linearity (R2) was found to be in the range of 0.9920 to 0.9974, and the percentage of relative standard deviation of the retention times (%RSD tR) were all below 0.2% (Table II).

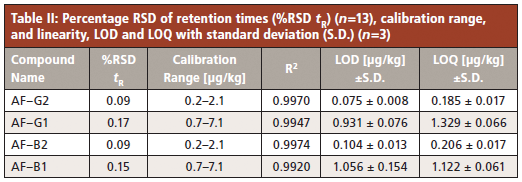
Immunoaffinity SPE purification has proven to be a very specific sample preparation method for ground hazelnut to achieve low LOD and LOQ levels. For the determination of LOD and LOQ values, one aliquot from each of the three sample extracts was pooled, diluted to a S/N ratio of 3 for LOD and S/N ratio of 10 for LOQ and injected three times. Figure 2 shows the resulting chromatogram of AF–B2 as one example. A full overview of the calibration results is given in Table II. Reliable quantification can be achieved with the applied method to safely determine the maximum levels of 5 µg/kg for AF–B1 and 10 µg/kg for the sum of all four aflatoxins, as regulated by the European Commission.

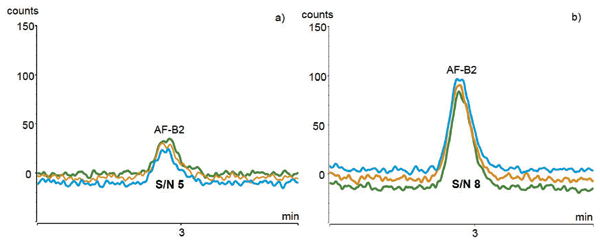
Figure 2: Exemplary chromatograms of aflatoxin B2 (triplicate injection) for the determination of a) LOD and b) LOQ.
After immunoaffinity SPE very pure extracts are obtained, as the chromatogram in Figure 3 demonstrates. The highly specific purification allows a separation of interfering matrix components, while enriching the target analytes. Some matrix peaks can be observed in the first 2 min, but no interferences in the target analyte region from 2 to 4 min are present. Neither the nonspiked sample extract nor the spiked (recovery) extract, where the standard solution was added before the sample preparation, show a peak in front of the toxin G2. In contrast, an impurity can be detected in the calibration standard, with the standard solution being added to the sample extract after the SPE cleanup (Figure 4). This clearly highlights that immunoaffinity SPE provides a distinct advantage in eliminating impurities present in the standard solution and in improving LOD and LOQ levels.


Figure 3: Overlay of spiked (recovery) sample (blue) and nonspiked (brown) hazelnut sample.
Furthermore, the overlaid chromatograms in Figure 4 show that no carryover in the blank injection was observed, even after injection the highest calibration point at a concentration of 2.1 µg/kg for AF–G2 and AF–B2 and 7.1 µg/kg for AF–G1 and AF–B1.

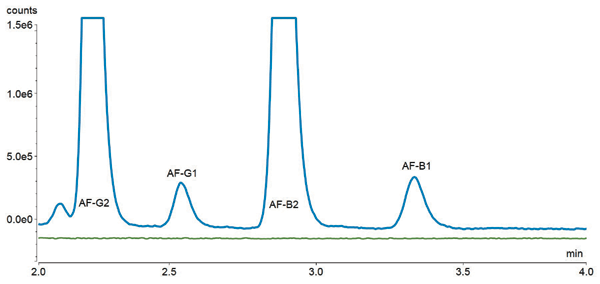
Figure 4: Zoomed overlaid chromatograms of processed sample spiked with the highest calibration concentration (blue) and consecutive blank injection (green).
Table III summarizes the quantitative results with recovery rates of each compound and calculated sample amounts (corrected by recovery rate).
The applied method provided excellent recovery rates for the toxins G2, B2, and B1 (95–100%) but somewhat inferior for AF–G1 with 72%. The shelf life of the cartridges specified by the manufacturer should be several months, if properly stored. The cartridges used in this study were close to their expiration date, possibly causing the lower AF–G1 recovery.

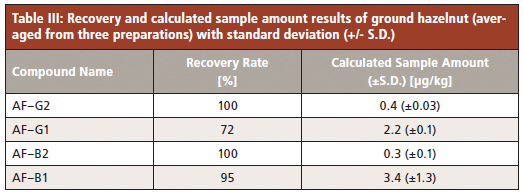
The major analyte of interest is the highly toxic AF–B1 and found to be present in the sample at an averaged concentration of 3.4 µg/kg (±1.3 µg/kg). The European Commission has set a limit of 5 µg/kg for this compound, which is just above the observed level. The sum of all four compounds should not exceed 10 µg/kg, and is calculated at an average of 6.3 µg/kg.
Conclusion
Aflatoxins are a group of mycotoxins, some of which are highly toxic to human health, and are known to cause various diseases. Therefore, the European Commission has set low µg/kg levels for aflatoxins in several foodstuffs used for direct consumption or as ingredients. Consequently, for their determination, a very sensitive and specific analytical method is required to enable trace-level detection of these analytes even in complex matrices. The combination of immunoaffinity SPE purification and enrichment with FLD detection without pre- or post- column derivatization offers a sensitive analytical method for the quantification of the aflatoxins G2, G1, B2, and B1 in ground hazelnuts. The applied method provides sufficient trace level detection performance down to 1 µg/kg for aflatoxins B1 and G1 and 0.1 µg/kg for B2 and G2, enabling aflatoxin analysis far below the tolerance levels defined by the European Commission. Good selectivity, linearity, and recovery for reliable quantitative results were observed, while the method run time of less than 4 min allows a high sample throughput.
References
(1) N.P. Gurav and S. Medhe, Anal Chem Ind J. 17(2), 126 (2018).
(2) B. Kabak, J. Sci. of Food and Agric. 89(4), 549-554 (2009).
(3) Commission Regulation (EC) No 1881/2006 of 19 December 2006: setting maximum levels for certain contaminants in foodstuffs, Annex section 2: mycotoxins (M5).
(4) N.W. Turner, H. Bramhmbhatt, M. Szabo-Vezse, A. Poma, R. Coker, S.A. Piletsky, Anal. Chim. Acta, 901, 12-33 (2015).
(5) M. Karsten, R. Swart, M. Mcleod, B. Murphy, S. Henderson, B. Richter, Fast end Effective Determination of Aflatoxins in Grains for Food Using Accelerated Solvent Extraction followed by HPLC, Chromatography-Foods-Beverages-Contaminants-Applications-Notebook 71476, 32-33 (HPLC 2008 presentation).
Sylvia Grosse, Mauro De Pra, and Frank Steiner are with Thermo Fisher Scientific, Germering, Germany. Please direct correspondence to sylvia.grosse@thermofisher.com.












Characterizing Plant Polysaccharides Using Size-Exclusion Chromatography
April 4th 2025With green chemistry becoming more standardized, Leena Pitkänen of Aalto University analyzed how useful size-exclusion chromatography (SEC) and asymmetric flow field-flow fractionation (AF4) could be in characterizing plant polysaccharides.




Investigating the Protective Effects of Frankincense Oil on Wound Healing with GC–MS
April 2nd 2025Frankincense essential oil is known for its anti-inflammatory, antioxidant, and therapeutic properties. A recent study investigated the protective effects of the oil in an excision wound model in rats, focusing on oxidative stress reduction, inflammatory cytokine modulation, and caspase-3 regulation; chemical composition of the oil was analyzed using gas chromatography–mass spectrometry (GC–MS).

