Essentials of LC Troubleshooting, Part II, Continued: More Misbehaving Retention Times


Some liquid chromatography (LC) troubleshooting topics never get old, because there are some problems that persist in the practice of LC, even as instrument technology improves over time. There are many ways for things to go wrong in an LC system that ultimately manifest as deviations from expected retention times. Lot-to-lot variability in column chemistry, effects of pressure on selectivity, and stationary phase loss are less frequently encountered causes of unexpected retention shifts, but ones that should be included in a comprehensive troubleshooting agenda.
In December of 2021, I kicked off an “Essentials of LC Troubleshooting” series of articles that is loosely organized around the well-known LCGC “Guide to LC Troubleshooting” wall chart (1) that hangs in many laboratories. Part II in the series (2) was focused on problems that lead to unexpected changes in retention times. For that installment, I focused on the most common problems, including changes in mobile phase flow rate, composition, pH, column temperature, and phenomena such as mass or volume overload and stationary phase dewetting. For this month’s installment, I am focusing on three problems that can all lead to unexpected changes in retention time, but in more complex ways than other simpler relationships, such as the effect of flow rate on retention time. As always, a wider and deeper understanding of factors that can affect retention time makes us more effective and efficient in the troubleshooting trade.
Topic #1: Lot-to-Lot Variations in Stationary Phase Chemistry
Some aspects of liquid chromatography (LC) technology have improved dramatically over the past few decades. For example, I don’t fret too much if I drop a modern column on the benchtop because I know that modern column packing methods make the particle bed quite resilient to such abuse.
This was not the case 40 years ago (at least this is what the veterans tell me), when columns were “dry packed,” and even relatively modest physical shocks to the column could irreparably ruin it. Similarly, whereas major fluctuations in column inlet pressure were commonplace with the relatively primitive pump designs in use 40 years ago, today’s modern pumps are remarkably good at delivering the mobile phase to the column in a way that is both accurate and very consistent.
Batch-to-batch or lot-to-lot reproducibility of LC stationary phases has also improved considerably over the years. However, this is—in my opinion—still a significant weakness in the LC enterprise where the reproducibility of a given method depends on tens, if not hundreds, of experimental variables. This is yet another example of a two-sided coin in LC—that, on one hand, we profit from the exquisite selectivity of LC columns and the fact that small changes in stationary phase chemistry can lead to meaningful improvements in resolution, and, on the other hand, this very sensitivity also means that manufacturers must have exquisite control over their stationary phase production processes if they are going to be able to deliver a highly reproducible product.
Figure 1 shows the difference in selectivity for two columns prepared with nominally identical stationary phases, but from two different manufacturing lots. Here, selectivity is defined as the retention factor of a given compound relative to the retention factor of toluene. We see that for most compounds, including neutrals and acids, the differences are small enough to be negligible for most practical purposes (<1%). However, for the two amines nortriptyline and amitriptyline, which are protonated and positively charged at pH 3, the differences are significant, on the order of 15%. A difference of this size could be enough to result in coelution, or even an elution order inversion on one column, even for two compounds that are completely resolved on the other column.

 relative to toluene (ktoluene). Chromatographic conditions: stationary phase is C18, mobile phase is 50:50 acetonitrile: 25 mM ammonium formate buffer, pH 3.2, temperature is 40 °C.")
FIGURE 1: Percent difference in selectivity for two columns with the same stationary phase, but from two different manufacturing lots. Selectivity is defined here as the retention factor of a given compound (kx) relative to toluene (ktoluene). Chromatographic conditions: stationary phase is C18, mobile phase is 50:50 acetonitrile: 25 mM ammonium formate buffer, pH 3.2, temperature is 40 °C.
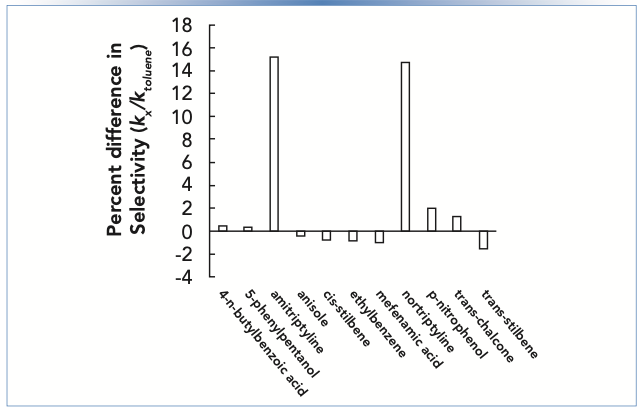
Experienced LC users are well aware that there can be significant lot-to-lot stationary phase variability, and they carefully evaluate this for any stationary phase that will be used in a validated method. Indeed, many column manufacturers sell method validation kits that are specifically assembled with columns drawn from different manufacturing lots. This makes evaluation of multiple lots easier for users.
If unexpected changes in retention time are observed after changing to a new column of the same stationary phase, one should seriously consider that the cause of the shift in retention could be because of lot-to-lot variability. If this is suspected, it is best to do a targeted comparison of retention using theold and new columns under the same conditions (such as same day, same instrument, same sample, and same mobile phase), so that the only variable in play is the stationary phase manufacturing batch.
Topic #2: Effect of Stationary-Phase Loss on Separation Selectivity
In the first installment discussing causes of changing retention times, I reviewed the primary mechanisms of stationary phase loss for reversed-phase (RP) columns (2). In the case of silica-based materials, hydrolysis of siloxane bonds anchoring stationary phase ligands to the silica surface is the major cause of phase loss at low pH, while, at high pH, dissolution of the underlying silica (and subsequent loss of ligands bonded to that silica) is the major cause of phase loss. One symptom of phase loss is a gradual change in retention on the timescale of days or months. The rate of change depends on a number of factors, including the specific chemistry of the stationary phase (for example, the hydrophobicity of the phase, steric protection of the siloxane bond, temperature, and mobile phase pH). If the rate of change is slow, and if the change in retention for all compounds in a separation is consistent, then such changes can be dealt with relatively easily, at least up to a point (for example, by adjusting peak integration windows within a predetermined range). However, the degree of change is often not the same for all compounds, and sometimes a significant compound dependence is observed.
Figure 2 shows selectivities measured using two columns from the same manufacturing lot of a short alkyl chain reversed-phase material, one new out of the box, and one that had been used for several months under mildly acidic conditions. The selectivities plotted are ratios of retention factors for the compounds shown relative to the retention factor for toluene. Above each pair of bars the percent change in selectivity for each compound is shown. We see that the percent changes are all negative, indicating a general loss in retention. However, the degree of change is compound dependent. The biggest difference in behavior we see with these compounds is between ethylbenzene and notriptyline. This difference, which is on the order of 5%, could easily result in a significant change in chromatographic resolution. In the best case two neighboring peaks would move apart and resolution would improve as the column changes. In the worst case, two peaks could converge, and enough resolution could be lost that accurate quantitation would no longer be possible.

FIGURE 2: Changes in selectivity of a reversed-phase column observed for different compounds after prolonged exposure to mildly acidic conditions. Selectivity is defined here as the retention factor of a given compound in 20:80 acetonitrile:buffer (kx,20) relative to the retention of toluene in 50:50 acetonitrile:buffer (ktoluene,50). Chromatographic conditions are the same as in Figure 1, except that the stationary phase is a short chain alkyl phase.
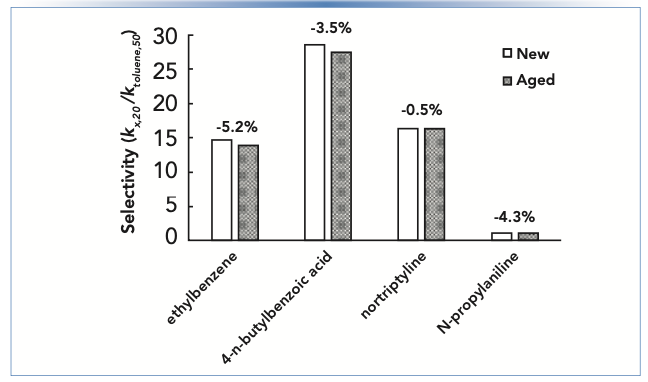
In situations where stationary phase aging is a suspected cause of unexpected retention time changes, historical data accumulated over time are invaluable. Such data can be obtained by routinely running system suitability tests (or similar) and used for diagnosing the situation and making a short list of potential causes to initiate a troubleshooting investigation.
Topic #3: Effect of Pressure (and Correlated Variables) on Separation Selectivity
Through my research group’s ongoing involvement in the development and use of the Hydrophobic Subtraction Model (HSM) (3), I am frequently asked whether one should expect columns packed with particles of different sizes, but the same stationary phase chemistry, to yield the same selectivity. This is clearly a very practically important question, especially currently given the diverse array of options for different particle sizes and morphologies (that is, fully porous vs. superficially porous) available to users. My answer is that, while there is no theoretical reason to think that the selectivity of a stationary phase should be dependent on particle size per se, what we observe in practice may be quite different.
One immediate concern is that two columns of the same diameter and length, and operated at the same flow rate, but packed with particles of different size will require different pressures to drive the fluid through them at the desired flow rate. In other words, the pressure drop across the two columns will be different, and thus pressures analytes experience inside the column will be different. Since the early work of McGuffin and Chen in this area (4), interest has steadily grown, with a recent report even suggesting the use of pressure as a variable to control selectivity during method development (5).
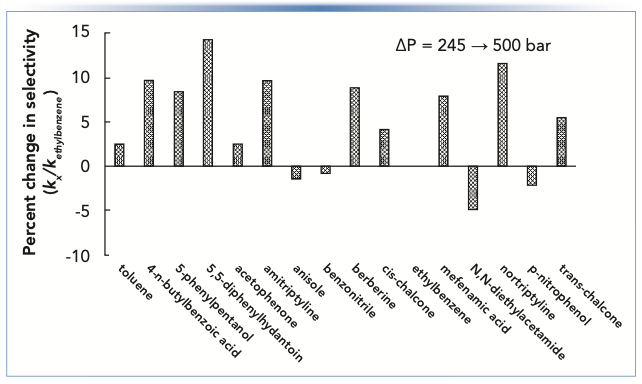
Figure 3 shows changes in selectivity for a C18 column (defined in this case as the retention factor of a given compound relative to the retention factor of ethylbenzene) that result from a change in the column midpoint pressure from 245 to 500 bar. We see that there is practically no effect on relatively small nonpolar molecules (that is, toluene, anisole, or benzonitrile, among others). However, the changes for larger and more polar molecules are significant with an average change of about 10%. Retention relative to ethylbenzene usually increases, but not always (6). As discussed in the Topic #2 section of this article, changes in selectivity on the order of 5–10% can cause the coelution of two compounds that were separated prior to the change. Thus, anything that changes the pressure that analytes experience inside the column can lead to such changes in selectivity. For example, in normal operation of a column, the pressure could change because of partial occlusion of a capillary or filter downstream from the column, though a change on the order of several hundred bar would be an extreme case. During method development, however, such changes in pressure could easily be encountered when changing from a column containing 5 μm particles to one containing sub-2-μm particles, particularly if the same flow rate is used for the two columns. In this case, one should not be surprised if significant selectivity changes are observed, even if the stationary phases are nominally identical for the two particle sizes.

FIGURE 3: Changes in selectivity of a reversed-phase column because of a change in pressure drop across the column. Chromatographic conditions: Stationary phase is C18, mobile phase is 50:50 acetonitrile:potassium phosphate buffer, pH 2.8, temperature is 40 °C. Pressure was adjusted by adding post-column restriction capillaries of different lengths (50 μm i.d.).
Summary
In this installment of “LC Troubleshooting,” I have discussed situations where the observed analyte retention is somehow different from what is expected or normal. This is a continuation of the discussion started on this topic in April of 2022. In this installment, we’ve addressed the effects that lot-to-lot stationary phase variation, stationary phase loss due to column aging, and pressure can have on separation selectivity, and therefore observed retention times. These effects are often more subtle compared to more obvious effects, such as pump failures that result in incorrect mobile phase composition, for example. Nevertheless, these more subtle effects are important parts of a comprehensive view of problems that can affect retention, which is a central metric of the performance of any high-performance liquid chromatography (HPLC) method. Understanding these details provides a good place to start our troubleshooting experience. Readers interested in learning more about commonly encountered problems and potential solutions are referred to the LCGC “Guide to LC Troubleshooting” wallchart.
References
(1) LCGC “Guide to LC Troubleshooting” Wallchart (n.d.). https://www.chromatographyonline.com/view/troubleshooting-wallchart
(2) Stoll, D. R. Essentials of LC Troubleshooting, Part II: Misbehaving Retention Times, LCGC N. Amer. 2022, 40 (4), 151–155. DOI: 10.56530/lcgc.na.fa1867h1
(3) Dolan, J. W.; Snyder, L. R. The Hydrophobic-Subtraction Model for Reversed-Phase Liquid Chromatography: A Reprise. LCGC N. Amer. 2016, 34 (9), 730–741.
(4) McGuffin, V. L.; Evans, C. E.; S.-H. Chen, S.-H. Direct Examination of Separation Processes in Liquid Chromatography: Effect of Temperature and Pressure on Solute Retention. J. Micro. Sep. 1993, 5, 3–10. DOI: 10.1002/mcs.1220050102
(5) Fekete, S.; Fogwill, M.; Lauber, M. A. Pressure-Enhanced Liquid Chromatography, A Proof of Concept: Tuning Selectivity with Pressure Changes and Gradients, Anal. Chem. 2022, 94, 7877–7884. DOI: 10.1021/acs.analchem.2c00464
(6) Kempen, T.; Stoll, D. R. Retention Factor is Independent of Pressure in LC, Right? LCGC N. Amer. 2021, 39 (10), 471–475.
About the Co-Authors

Kathryn Cash (left) and Ezedin Seid (center) are undergraduate researchers, and Tina Dahlseid (right) is a Staff Scientist, all working with Dwight Stoll's research group at Gustavus Adolphus College, in St Peter, Minnesota.

About the Column Editor

Dwight R. Stoll is the editor of "LC Troubleshooting." Stoll is a professor and the co-chair of chemistry at Gustavus Adolphus College in St Peter, Minnesota. His primary research focus is on the development of 2D-LC for both targeted and untargeted analyses. He has authored or coauthored more than 75 peer-reviewed publications and four book chapters in separation science and more than 100 conference presentations. He is also a member of LCGC's editorial advisory board. Direct correspondence to: LCGCedit@mmhgroup.com



















Polysorbate Quantification and Degradation Analysis via LC and Charged Aerosol Detection
April 9th 2025Scientists from ThermoFisher Scientific published a review article in the Journal of Chromatography A that provided an overview of HPLC analysis using charged aerosol detection can help with polysorbate quantification.


Removing Double-Stranded RNA Impurities Using Chromatography
April 8th 2025Researchers from Agency for Science, Technology and Research in Singapore recently published a review article exploring how chromatography can be used to remove double-stranded RNA impurities during mRNA therapeutics production.

