The Early Development of Size Exclusion Chromatography: A Historical Perspective
LCGC Europe
In this month's column, Howard Barth traces the early development of SEC
Size exclusion chromatography (SEC) is a well-established high performance liquid chromatography (HPLC) method for separating macromolecules according to their hydrodynamic volume. When SEC was first recognized 60 years ago by biochemists it was an instant success, and was mainly used for analysing complex biological samples, desalting biological solutions and for preparative separations of biopolymers. Some years later, cross-linked polystyrene packings were introduced to determine molecular weight distributions of synthetic polymers. With time, SEC evolved to become the mainstay of practically all laboratories concerned with macromolecular characterization. In this month's History of Chromatography instalment, Howard Barth traces the early development of SEC.
Separation by size exclusion chromatography (SEC) is governed solely by the hydrodynamic size and shape of macromolecules relative to the size and shape of the pores of the column packing. Provided that the proper mobile phase is selected, the entire polymer sample will elute from the column within a defined elution volume region dictated by the porosity of the packing and column dimensions. The elution order of a homologous series of components is always large molecules first, followed by smaller-sized components, which aids data interpretation.
SEC, when compared to high performance liquid chromatography (HPLC), requires no gradient elution and, to a first approximation, no temperature control. Method development is usually straightforward: for the analysis of biopolymers, an aqueous buffer is used, while for synthetic polymers, an organic solvent is used for the mobile phase.


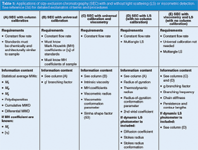
Table 1: Applications of size exclusion chromatography (SEC) with and without light scattering (LS) or viscometric detection. See reference (24) for detailed explanation of terms and procedures.
With this simple system, a wealth of information can be obtained about the properties and characteristics of macromolecules (Tables 1 and 2). Because of the usefulness of this technique, especially for the analysis of complex samples with a wide molecular weight range, most HPLC laboratories have a dedicated SEC instrument to aid in problem-solving.


Table 2: Conventional and unconventional size exclusion chromatography applications without column calibration.
It took nearly 50 years from Tswett's momentous discovery in 1906 of column liquid chromatography before SEC was first recognized, quite by accident, as a new separation mode. Once it was introduced to the scientific community, it became an essential technique for the life sciences and polymer science.

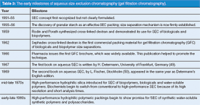
Table 3: The early milestones of aqueous size exclusion chromatography (gel filtration chromatography).
In this article, we will trace the early development of SEC from its inception to its acceptance as a modern analytical technique for biopolymer and synthetic polymer characterization. Some of the significant SEC milestones that will be covered are summarized in Tables 3 and 4.


Table 4: The early milestones of non-aqueous size exclusion chromatography (gel permeation chromatography).
In the Beginning
Gel "Filtration" Chromatography (GFC): In the early 1950s, investigators (1,2) had noted that neutral small molecules and oligomers eluted on the basis of decreasing molecular weight from columns packed with cross-linked polystyrene ion-exchange resins. Intrigued by this separation mode, Deuel (3) and Lindquist (4) reported similar results for the elution of low-molecular-weight solutes using polysaccharide packings, that is, cross-linked galactomannan and starch granules.
In 1955–56, two British biochemists, Lathe and Ruthven (5,6), used conventional open columns packed with swollen starch granules to study the elution properties of low- and high-molecular-weight polysaccharides and proteins. To their surprise, higher molecular weight compounds eluted ahead of lower molecular weight components, in agreement with earlier work (1–4). Because of Lathe and Ruthven's more thorough investigations, they are credited with performing the first biopolymer size separation (see Figure 1). Although they correctly described the separation as being caused by "partial or size-restricted penetration of solute molecules into the swollen gel particles", Lathe and Ruthven incorrectly called the process "gel filtration", which implies that the high-molecular-weight components are physically retained on the column.

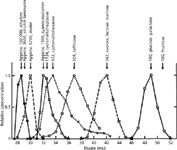
Figure 1: First published SEC composite chromatogram of polysaccharides, oligosaccharides, carbohydrates and vitamin B12 using swollen potato starch (75 g filled to 56 cm) and a buffer for the mobile phase at 4 mL/h. Fractions were collected and analysed via UVâvis spectrophotometry using post-column derivatization. The following elution volumes are estimated from the data provided: V0 â 28 mL; Vi â 46 mL; Vt â 74 mL; Vi /V0 â 1.6 which is satisfactory. Since N â 1000 plates (glucose) and H â 600 μm, column efficiency is at least 30x lower than a typical HPLC column, which is a reasonable value considering this is a conventional open-column separation. Total elution time for this system is about 20 h. Adapted and reproduced with permission from G.H. Lathen and C.R.J. Ruthven, (1956), Biochemical Journal, 62, 665â674. © the Biochemical Society.
The next milestone occurred in 1959, when Per Flodin, a biochemist with Pharmacia Pharmaceutical Company (Sweden), in collaboration with Jerker Porath, synthesized a series of cross-linked dextran packings with different pore sizes, and successfully demonstrated size separation of peptides and oligosaccharides (7–9). Since Pharmacia had considerable experience with the production and chemistry of dextran, which they used as a blood plasma substitute for transfusions, the company was able to quickly produce and market cross-linked packings of different porosities in the early 1960s. The packing was trademarked Sephadex, an acronym taken from the first few letters of separations Pharmacia dextran. Sephadex sales soared, and soon most biochemistry laboratories used GFC. Within a short time, other types of cross-linked gels were released by Pharmacia.
Apart from being used for estimating molecular weights of biological components from elution time comparisons to molecular weight standards, Sephadex was primarily used in place of dialysis for desalting or exchanging electrolytes in biopolymer preparations, or for isolating and purifying biopolymers and other biologicals. (It seemed that most biochemical laboratories at the time were situated next to slaughterhouses to ensure a steady stream of starting material.)
Gel Permeation Chromatography (GPC): Motivated by the early success of gel filtration chromatography, polymer scientists in the late 1950s and early 1960s focused their efforts on the application of hydrophobic packings for the SEC of synthetic polymers, at the time referred to as gel permeation chromatography (GPC). The concept of molecular weight separation of nonpolar molecules was first demonstrated when Brewer (10, 11) used swollen rubber granules for the separation of a homologous series of hydrocarbons. Determann et al. (12) evaluated cross-linked poly(methyl methacrylate) for SEC of oligostyrenes.
In 1962, John Moore (13–15) of Dow Chemical Company (Midland, Michigan, USA) produced a series of cross-linked polystyrene resins of known porosities and particle sizes for the SEC of synthetic polymers. For the first time, the molecular weight distribution of any polymer soluble in toluene could be obtained within several hours compared to weeks or months with conventional fractionation and procedures. Soon after the method was published, polymer laboratories around the world clamoured for these packings and associated GPC instrumentation.
Dow Chemical recognized the practicality and importance of GPC to their polymer business and licensed the packing synthesis and instrumentation to Waters Associates, a consulting independent laboratory located near Boston, USA. James Waters, its founder, had rented the basement of a former police station for this new business venture, and had set up polymerizers on the first floor in an area that was formerly a women's jail. Waters soon supplied packed columns and complete GPC systems for room temperature; high-temperature (135 °C) SEC for polyolefins; and large-scale preparative separations. Waters Associates became the GPC counterpart of Pharmacia's GFC venture. Waters Associates was the only source of GPC packings and instrumentation for many years.
Packings
Initially SEC packings consisted of either cross-linked polysaccharides for aqueous SEC or cross-linked polystyrene for non-aqueous SEC separations, as listed in Tables 3 and 4. Packings for GFC were developed by Pharmacia (Sweden), Bio-Rad Laboratories (USA), LKB Produkter (Sweden) and E. Merck (Germany). Waters Associates (Waters Corp., Milford, Massachusetts, USA), which licensed the polystyrene polymerization technology from Dow, and held the "GPC" name as their trademark, was the sole producer of Styragel packings.
In the early 1970s, reduced particle size, porous silica was shown to have extremely high plate counts for HPLC separations, including SEC, because of short diffusion paths travelled by solutes. Over time, biochemists switched from large-diameter cross-linked polysaccharide packings to high-performance silica-based packings and then on to hydrophilic polymer packings for aqueous SEC as they became available in later years. Small diameter cross-linked polystyrene packings, which were made rigid and non-compressible, became available in the mid-1980s.
Companies that helped launch and promote the early development of high-performance SEC packings were: Varian Inc., BioRad Inc., Pharmacia, Waters Associates, DuPont Instruments, Toyo Soda (now Tosoh Bioscience), Showa Denko, Co., Synchrom Inc. and E. Merck. Although controlled-pore glass (CPG) was not a high-performance packing, it was available in very large pore sizes with narrow pore-size distributions. Of these companies, Waters, Tosoh Bioscience (King of Prussia, Pennsylvania, USA) and Showa Denko (Kawasaki, Kanagawa, Japan) remain, and continue to advance, the technique. Other major SEC column suppliers include Agilent Technologies (Wilmington, Delaware, USA), Polymer Laboratories (Church Stretton, Shropshire, UK), PSS Polymer Standards Service (Mainz, Germany), Phenomenex (Torrance, California, USA), Jordi Associates (Bellingham, Massachusetts, USA), BioChrom Labs (Terre Haute, Indiana, USA), Eprogen (Downers Grove, Illinois, USA), Sepax Technologies (Newark, Delaware, USA) and YMC (Allentown, Pennsylvania, USA).
Instrumentation
Pumps: Because of the nature of SEC calibration, in which logM is plotted against elution volume, small flow rate deviations cause large molecular weight errors, especially when calculating average molecular weights of polydisperse samples. However, for proteins and nucleic acids, which are mostly monodisperse, biochemists used SEC for determining molecular weights at peak maxima, which was less dependent on flow rate variations. It is of interest to note that the most popular uses of GFC were desalting, electrolyte exchange or biopolymer purification, which do not necessarily require high-precision flow rate.
The most common pumping systems for GFC were peristaltic pumps or motor-driven glass syringes, which are still in use. Gravity-induced flow with open-column chromatography was widely used. To help control flow with gravity feed, a simple device called the Mariotte flask is often used. The Mariotte flask, which serves as the mobile-phase reservoir, has a tube inserted for draining mobile phase onto the top layer of the packed column. The flask also has a capillary tube with one end kept below mobile-phase level and the other end connected to the outside for admitting air into the flask via a steady stream of bubbles. Mobile-phase flow rate is controlled by adjusting the stream of air bubbles, and the flow rate determined by measuring mobile-phase depletion in the flask.
When determining molecular weight averages of polydisperse samples, as in the case of GPC, precise flow rate control is required. Most pumps at the time were industrial single- or dual-piston pumps with modified seals for chemical inertness; metering pumps from Milton Roy were commonly used with conventional column LC.
To monitor flow rate throughout a GPC run, a custom-made siphon "dump" was used up until the early 1980s. This device was placed at the end of the column. The siphon, which had a capacity of 1-, 2- or 5-mL, would fill to volume and then dump its contents, producing a signal at each volume increment. The average flow rate throughout the run could be computed.
Detectors: When GFC was introduced in the 1950s, flow-through detectors were not available. Instead, manual or automated fraction collection was accomplished with subsequent UV measurements. For monitoring oligosaccharides, polysaccharides or lipids, fraction collection with post-column derivatization and subsequent UV–vis spectrometry was required. Obviously, only a few data points across a peak could be taken, and a great deal of peak broadening was introduced, as seen in Figure 1. For coloured solutes, migration through glass columns could be followed visually.
Data Acquisition and Processing: Although it is difficult to imagine a time before dedicated computers, it did exist! Since cross-linked polysaccharide gels were soft and compressible, low pressures or flow rates were required. As such, a single GFC run would take approximately 12 h, and an additional one or two days to analyse collected fractions and plot the results. The total time for a single GFC analysis would be approximately 1.5 to 2.5 days, or close to a week for a duplicate run.
Cross-linked polystyrene packings for GPC were more robust, so higher flow rates could be used. Depending on the number of Styragel columns that were in series, a single run could take about 1.5 h, with an additional 2.5 h to manually measure peak heights off a chromatogram, tabulate the data and then calculate average molecular weights and graph cumulative and differential molecular weight distributions. Total time would be roughly 4 h per sample. If a large-frame computer were available with data entry using punched tape or cards, total analysis time could be reduced to about 2.5 h per sample. Tedious, yes, but SEC was much faster than the weeks required for classical fractionation with subsequent osmometry and light scattering measurements of every fraction collected.
SEC Elution Behaviour
In the early history of SEC, experiments had confirmed that log M vs. elution volume has a negative, nearly linear slope. Each polymer type was found to have a characteristic calibration slope and intercept. Based on these observations, the SEC elution behaviour of macromolecules is

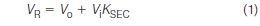
where VR is the elution volume of a given component, Vo is the interstitial volume of the packed column, Vi is the total pore volume of the packed column and KSEC is the distribution coefficient of the SEC separation process. The maximum or total elution volume Vt of a packed column is

and the distribution coefficient (or partition coefficient at ideal conditions) of a solute is

where c1 and c2 are the average solute concentrations in the pore volume and in the interstitial volume, respectively. The distribution coefficient ranges from zero to unity. Macromolecules too large to access the pore volume will elute at V0. Small molecules that can freely diffuse into the pores of the packing will have an elution volume of Vt.
Thermodynamics of SEC
It is instructive to examine the thermodynamics of a separation to provide additional insight into the nature of the process. The distribution coefficient, Kt, of a system at equilibrium and infinite dilution is
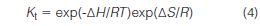
where ΔH is the change in enthalpy when a solute is transferred from phase 2 to 1, ΔS is the entropy change when the solute diffuses from phase 2 to 1, R is the gas constant and T is the absolute temperature of the system. In SEC, it is critical that macromolecules do not interact energetically with the outer surface or pore walls of the packing, thus

and equation (4) becomes
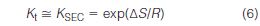
where ΔS is the conformational entropy change of a macromolecule when it diffuses from one phase to another phase, whereby the concentration gradient of the solute is the driving force.
Provided that we keep ΔH = 0 by the judicious selection of packing material and mobile phase composition, the separation will depend only on the size and shape of macromolecules with respect to the physical characteristics of the pore structure. An examination of equation (6) reveals that the SEC process is independent of temperature, unlike all other chromatographic separations.
Please note that even if 0 ≤ KSEC ≤ 1, an SEC separation may not necessarily be governed strictly by molecular size. This can happen if the ionic strength of the mobile phase is insufficient to prevent the occurrence of ion exclusion or ion inclusion when analysing polyelectrolytes (16). Also, if the injected polymer concentration is too high, concentration effects (17), such as macromolecular crowding or viscous "fingering", may distort peak shapes or artificially increase elution volume.
The thermodynamic basis of SEC was confirmed by Casassa in a series of theoretical papers published from the mid-1960s to early 1970s (18–23). In these studies, he derived distribution coefficients for linear and branched polymers and different pore geometries using statistical mechanical calculations. His results were in good agreement with experimental data, especially when a slab-shaped pore geometrical model was compared to published data.
Ultrahigh-Molecular-Weight Polymers
There have been other SEC separation mechanisms proposed over the years (24, 25), but they have not supplanted the thermodynamic model. Discrepancies were noted for SEC of ultrahigh-molecular-weight (UHMW) samples, but these deviations could be explained by the onset of other separation mechanisms, such as polymer chain deformation, which orientates the chain in the direction of flow (26); hydrodynamic chromatography (27, 28), in which macromolecules elute earlier than predicted by SEC; or slalom chromatography (29, 30), where macromolecules elute later than expected.
SEC of UHMW polymers is fraught with other difficulties besides mechanistic uncertainty. These materials can easily shear degrade during sample preparation, as well as during column elution (31). Because of their extremely low diffusion coefficients, UHMW samples undergo significant peak broadening, obfuscating data interpretation (17). Furthermore, depending on the polymerization process, UHMW samples may contain intractable gels, entangled networks or lightly cross-linked chains that can accumulate within the column and be filtered out, altering the molecular weight distribution profile. UHMW samples may require alternative low-shear rate methods of characterization, such as analytical ultracentrifugation, field flow fractionation or dynamic light scattering.
Column Resolution
SEC resolution or performance depends on the slope of the calibration, as well as column efficiency. To take these parameters into account, Yau et al. (32) formulated a specific resolution equation
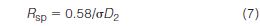
where D2 is the slope of the calibration plot and σ is the peak-width standard deviation of a monodisperse polymer. To compare resolution among columns of different lengths, L, Yau normalized equation (7) with respect to column length
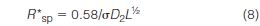
where R*sp is the specific resolution for a 1-cm column. The benefit of this equation is that it can be used for comparing column resolution or performance among columns of different lengths. In fact, any of the series of resolution equations derived by Yau (32) provide the user with important performance parameters that can be readily calculated and used for selecting the best column or column set.
Peak Broadening
Peak broadening in SEC can lead to serious errors when determining average molecular weights of polydisperse samples. Essentially, peak broadening will cause the high-molecular-weight end of a distribution to diffuse into the earlier eluting region, and the low-molecular-weight end to diffuse into the later elution zone. The net result is an artificially broadened sample, or as Yau (24) explains, peak broadening rotates the calibration plot counter-clockwise. This means that if a multipoint calibration is used to calculate average molecular weights, the weight-average molecular weight, Mw, will be overestimated, the number-average molecular weight, Mn, will be underestimated, and the polydispersity will be overestimated by amounts governed by the degree of peak broadening.
A key development in SEC was the derivation of useful equations by Yau (24) to estimate molecular weight errors introduced by peak broadening

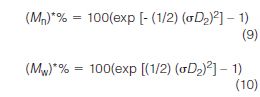
Here (Mn)*% and (Mw)*% are the estimated relative (percent) errors encountered when determining number- and weight-average molecular weights from a multipoint calibration curve. These equations and others (24) can be used for estimating errors caused by peak broadening and taking the necessary steps to lower σD2 to an acceptable value. This reference also contains a wealth of information on recommended peak broadening corrections.
References (33) and (51), although outdated, are American Society for Testing and Materials (ASTM) committee reports that describe a simple approach for correcting asymmetrical and symmetrical peak broadening, which incidentally had undergone round-robin testing. Unfortunately, computer software with peak broadening corrections has not been readily available, so the majority of SEC practitioners tend to use high efficiency columns; low flow-rate; low injection concentration and volume; and elevated column temperature, and hope for the best.
Except for the broad-molecular-weight-standard approach (17, 24), all other multipoint calibration methods, including universal calibration, with or without on-line viscometry, are subject to errors caused by peak broadening. It should be noted that even if an on-line light scattering detector is employed, peak broadening will cause errors in polydispersity and all molecular-weight averages, except for Mw (34). The only SEC detector that would be free of peak broadening errors is electrospray ionization (ESI) or matrix-assisted laser desorption ionization (MALDI) mass spectrometry (MS), provided that ion intensity can be used to determine the concentration of eluting components.
Column Calibration
Since SEC is a relative, not an absolute method, a great deal of effort has gone into devising accurate calibration approaches. The most popular methods have been: 1. calibrating with a set of primary polymer calibrants of narrow polydispersities; 2. calibrating with one or more polydisperse polymer standards of known Mn and Mw; 3. calibrating with any set of monodisperse polymer standards as long as the intrinsic viscosity or Mark-Houwink coefficients of the standard and sample are known, as discussed below.
In 1966, Benoit and co-workers (35) demonstrated that if log [η]M is used instead of log M, the calibration plots of chemically and architecturally different polymers will merge into a single "universal" calibration plot.
The reason that [η]M generates the same calibration for different types of polymers is because this product is equal to the molecular hydrodynamic volume Vh of a polymer of molecular weight M in a given solvent (that is, mobile phase) and column temperature:

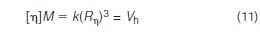
A universal calibration is generated for an SEC system by injecting a series of monodisperse polymer standards; corresponding elution volumes are plotted against log[η]M, where [η] of each standard can be obtained from the vendor, measured in the laboratory or computed from the Mark-Houwink equation

using literature values of Mark-Houwink coefficients K' and a.
By using universal calibration, the sample molecular weight at each elution volume increment designated by subscript i is

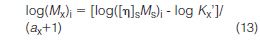
where subscripts x and s refer to sample and universal calibration standard, respectively. Equation (13) can be used to determine the molecular weight of any polymer at each elution volume increment provided that the Mark-Houwink coefficients, a and K', are known for the sample.
This discovery of universal calibration by Benoit is remarkable for two reasons: Firstly, universal calibration confirms that SEC separation is based on hydrodynamic volume differences; and secondly, universal calibration allows us to obtain molecular weight data for any polymer sample, as long as appropriate Mark-Houwink coefficients are available.
For an approximate 20-year period, universal calibration had not been as widely used as anticipated because of the lack of reliable Mark-Houwink coefficients. Furthermore, many laboratories were hampered because of the unavailability of universal calibration software for data acquisition and processing. To meet these challenges, investigators began to design and write their own software and produce flow-through viscometers. These developments will be discussed next.
Early SEC Detectors
A detailed review of all early SEC detectors, including the experimental ones that were not commercially successful, is beyond the scope of this article. In addition, HPLC spectroscopic detectors, such as MS, nuclear magnetic resonance (NMR), Fourier-transform infrared spectrometry (FT-IR), electrochemical and other property-specific detectors will not be covered since they all are part of the history of HPLC, not SEC. Instead the focus of this section is on those detectors that have had the greatest impact on SEC.
Differential Refractometer: In 1962, at the request of John Moore at Dow Chemical, James Waters produced the first successful flow-through differential refractometer (DRI) for SEC with a specified cell volume of <100 μL. The DRI was a major breakthrough for SEC and still remains the ideal SEC detector except for oligomers below a degree of polymerization of approximately 10–20.
The detector Waters developed was the "deflection" type; remarkably, this type is still the DRI of choice for SEC, with, of course, advanced electronic and optical systems, improved temperature stability and at least a 12-fold decrease in cell volume. Key to its success is its original diagonal-cell design, with a flow-reference cell and position-sensitive photocell (recently upgraded to a diode-array configuration). The current DRI instruments have reduced baseline drift and short-term noise that originate from temperature and flow-rate fluctuations.
Viscometry: As discussed in the preceding section, universal calibration was the driving force behind the development of on-line viscometers. If the intrinsic viscosity of the sample ([η]x)I could be determined at each elution volume increment, then the molecular weight of the sample (Mx)i can be readily calculated with the aid of a universal calibration plot without the need for Mark-Houwink coefficients:

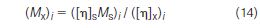
The first experimental "on-line" viscometer used for this purpose was constructed by Meyerhoff (36) in 1968 and consisted of a bank of six semi-automatic Ostwald-type viscometers arranged in a circular fashion. One viscometer, presumably at the centre, continuously measures flow time t0 of the mobile phase collected from the reference side of a DRI. Eluent from the sample side of the DRI flows in a sequential manner into each of the other viscometers with a 60 s interval between each. These viscometers are used to determine the capillary flow time, t, of the eluting polymer. For each data point, intrinsic viscosity measurements are determined using the following equation in which the concentration, c, is measured by the DRI
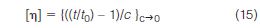
It is of interest to note that this rather complicated instrument can probably be duplicated using a present-day automatic viscometer, equipped with an HPLC fraction collector and automatic sampler.
In the mid-1980s, sensitive differential pressure transducers became available in different configurations and sizes for measuring the pressure drop across capillary tubes. Based on Poiseuille's equation, the intrinsic viscosity [η] at each elution volume increment i can be determined from the pressure drop, ΔP, that develops across the capillary when the polymer elutes, normalized with respect to the pressure drop, ΔP0, when just the mobile phase passes through the capillary
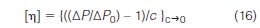
In this capillary arrangement, the pressure drop of pure mobile phase, ΔP0, must be determined before or after the polymer elutes.
Rather than using a single capillary, which can be quite noisy, a reference capillary is added, which not only monitors ΔP0, but also serves to cancel noise caused by temperature and flow-rate fluctuations. Currently, different models of commercial viscosity detectors are available, each having their own capillary configuration (34, 37–40).
Light Scattering: Rayleigh light scattering is "the gold standard" of weight-average molecular weight measurements. This well-known method can also determine z-average radius of gyration and the second virial coefficient of polydisperse polymer samples; therefore it was only a matter of time before a commercial instrument would be available. Kaye and co-workers (41–43) are credited with the first low-angle light scattering photometer for SEC, which was commercialized by Chromatix (USA) (44) in the mid-1970s (model KMX6). By the beginning of 1980 (45), a second Chromatix model dedicated to SEC was introduced (model CMX100). Unfortunately, Chromatix, which was sold to LDC (USA), no longer exists. Toya Soda (now Tosoh Bioscience, Japan) also issued a low-angle unit for SEC in the early 1980s, but eventually stopped making it. The two Chromatix instruments measured scattered light at low angles, which precluded molecular size (radius-of-gyration) measurements.
Next to appear in the mid-1980s was a flow-through multiangle light scattering instrument (DAWN model F) by Wyatt Technology Corporation (Santa Barbara, California, USA) that was capable of measuring up to 18 angles simultaneously using an array of detectors surrounding the flow cell (34,46,47). With this instrument, the radius-of-gyration could be determined, as well as the molecular weight at each elution volume increment.
A flow-through, dynamic light scattering (DLS) detector briefly appeared on the market in the mid-1980s from Oros Instruments in the UK (model 801). Although this DLS instrument had a large cell volume and low sensitivity, it helped to pave the way for later, improved models from Brookhaven Instruments (Holtsville, New York, USA), Precision Detectors (Bellingham, Massachusetts, USA) and Wyatt Technologies.
In the late 1980s to early 1990s, Wyatt continued to release new versions of SEC light-scattering photometers with updated electronics, optics and software. More recently, multiangle light scatter instrumentation became available from PSS Polymer Standards Service and Brookhaven Instruments.
Researchers in the late 1980s used light scattering and DRI detectors in conjunction with on-line viscometry to generate accurate branching and molecular conformation information, in addition to molecular weight and size data. A similar detector configuration, called the "triple detector", was initially available from Viscotek (Houston, Texas, USA) to correct their right-angle light-scattering data for anisotropy (34). At the time of writing, Precision Instruments and Malvern (Worcestershire, UK) market this three-detector system.
Conclusions
After the discovery of gel filtration chromatography in 1955, biochemists looked upon GFC as a revelation, at a time when many laboratories were involved with laborious preparative separations and purification of small quantities of biologically active biopolymers from kilograms of material collected from the neighbourhood slaughterhouse.
In 1966, as GPC instrumentation and packings were being made available to polymer scientists, there was a tremendous growth of synthetic polymers. For example, synthetic polymer production in the United States more than doubled each year from 1957 to 1973. According to Brydson (48), this "spectacular growth of polymers", was partly a result of the emergence of new polymers and applications; improved quality of existing polymeric material; and a greater than ever understanding of polymer processes, properties and characteristics.
It is obvious to this author that GPC played a significant role in guiding polymer technology through these three critical areas.
Howard G. Barth retired from the DuPont Company as senior research associate, where he was involved with SEC, HPLC and polymer characterization. He was appointed associate editor of the Journal of Applied Polymer Science, and also book series editor of Springer's Laboratory Manuals in Polymer Science. He was founding editor and editor-in-chief of the International Journal of Polymer Analysis and Characterization, and was chair for many years of the International Symposium on Polymer Analysis and Characterization. He works as a consultant in chromatography, analytical chemistry and technical editing and writing. Please direct correspondence to: howardbarth@gmail.com.
References
(1) H. Deuel, J. Solms and L. Anyus-Weisz, Helv. Chim. Acta, 33, 2171 (1951).
(2) R.M. Wheaton and W. C. Baumann, Ann. New York Acad. Sci., 57, 159 (1953).
(3) H. Deuel and H. Neukom, Adv. Chem. Ser., 11, 51 (1954).
(4) B. Lindqvist and T. Storgaards, Nature, 175, 511 (1955).
(5) G.H. Lathe and C.R.J. Ruthven, Biochem. J. 60, 34 (1955).
(6) G.H. Lathe and C.R.J. Ruthven, Biochem. J. 62, 665 (1956).
(7) J. Porath and P. Flodin, Nature 183, 1657 (1959).
(8) J. Porath, Clin. Chem. Acta, 4, 776 (1959).
(9) W. Bjork and J. Porath, Acta Chem. Scand., 13, 1256 (1959).
(10) P.J. Brewer, Nature, 188, 934 (1960).
(11) P.J. Brewer, Nature, 190, 625 (1961).
(12) H. Determann, G. Luben and Th. Wieland, Makromol. Chem., 73, 168 (1964).
(13) Anon., Chem. Eng. News, 40(51), 43 (1962).
(14) J.C. Moore, J. Polym. Sci., A2, 835 (1964).
(15) J.C. Moore, J. Polym. Sci., C(21), 1 (1968).
(16) H.G. Barth, J. Chromatogr. Sci., 18, 409 (1980).
(17) S. Mori and H.G. Barth, Size Exclusion Chromatography (Springer Verlag, Berlin, 1999).
(18) E.F. Casassa, J. Polym Sci., B5, 773, (1967).
(19) E.F. Casassa, In Characterization of Macromolecular Structure; D. McIntyre, ed., (National Academy of Science, Washington, D.C. 285,1968).
(20) E.F. Casassa, Polym. Preprints, ACS Div. Polym. Chem., 9, 565 (1968).
(21) E.F. Casassa and Y. Tagami, Macromolecules, 2, 14 (1969).
(22) E.F. Casassa, Sep. Sci., 6, 305 (1971).
(23) E.F. Casassa, J. Phys. Chem., 75, 3929 (1971).
(24) A.M. Striegel, W.W. Yau, J.J. Kirkland and D.D. Bly, Modern Size Exclusion Chromatography, 2nd Ed. (Wiley, New York, 2009).
(25) J.H. Aubert and M. Tirrell, Sep. Sci. Technol., 15, 123 (1980).
(26) A.M. Striegel, J. Liq. Chromaogr. Rel. Technol., 31, 3105 (2008).
(27) S.S. Huang, In: Handbook of Size Exclusion Chromatography and Related Techniques, 2nd Ed., C.-S. Wu, ed., p.677 (M. Dekker, New York, 2004).
(28) G. Stegeman, J.C. Kraak and H. Pope, J. Chromatogr., 550, 721 (1991).
(29) Y. Liu, W. Radke and H. Pasch, Macromolecules, 38, 7476 (2005).
(30) C. DeLong and D.A. Hoagland, Macromolecules, 41, 4887 (2008).
(31) H.G. Barth and F.C. Carlin, J. Liq. Chromatogr., 7, 1717 (1984).
(32) W.W. Yau, J.J. Kirkland, D.D. Bly and H.J. Stoklosa, J. Chromatogr., 125, 219 (1976).
(33) ASTM D-3536-76, Standard Method for Molecular Weight Averages and Molecular Weight Distribuion of Polystyrene by GPC.
(34) C. Jackson and H.G Barth, In: Handbook of Size Exclusion Chromatography and Related Techniques, 2nd Ed., C.-S. Wu, ed., p.99 (M. Dekker, New York, 2004).
(35) Z. Grubisic, P. Rempp and H. Benoit, J. Polym. Sci., Polym. Lett. Ed., 5, 753 (1967).
(36) G. Meyerhoff, Makromol. Chem., 118, 265 (1968).
(37) M.A. Haney, J. Appl. Polym. Sci., 30, 3023 (1985).
(38) M.A. Haney, J. Appl. Polym. Sci., 30, 3037 (1985).
(39) J.L. Ekmanis and R.A. Skinner, J. Appl. Polym. Sci., Symp. Ed., 48, 57 (1991).
(40) W.W. Yau, S.D. Abbott, G.A. Smith and M.Y. Keating, ACS Symp. Ser., 352, 80 (1987).
(41) W. Kaye, Anal. Chem., 45, 221A (1973).
(42) W. Kaye and A.J. Havlik, Appl. Opt., 12, 541 (1973).
(43) A.C. Oano and W. Kaye, J. Polym. Sci., Part A1, 12, 1151 (1974).
(44) M.L. McConnell, Am. Lab., 10(5), 63 (1978).
(45) R.C. Jordon, J. Liq. Chromatogr., 3, 439 (1980).
(46) P.J. Wyatt, C. Jackson and G.K. Wyatt, Am. Lab., 20(5), 86 (1988).
(47) P.J. Wyatt, D.L. Hicks, C. Jackson, and G.K. Wyatt, Am. Lab., 20(6), 108 (1988).
(48) J. Brydson, Plastics Materials, 7th Ed., Chpt. 1 (Butterworth-Heinemann, Oxford, 1999.)
(49) H. Determann, Gel Chromatograhy, 2nd Ed. (Springer-Verlag, New York, USA, 1969).
(50) L. Fischer, Gel Filtration Chromatography, 2nd Ed. (Elsevier, Amsterdam, North Holland, 1980).
(51) ASTM D-3593-80, Standard Method for Molecular Weight Averages and Molecular Weight Distribution of Certain Polymers by GPC Using Universal Calibration.
(52) W. W. Yau, J. J. Kirkland and D. D. Bly, Modern Size Exclusion Chromatography (Wiley, New York, USA, 1979).
(53) C.R. Cantor and P.R. Schimmel, Biophysical Chemistry, Part II (W.H. Freeman, New York, USA, 1980).
















Evaluating Body Odor Sampling Phases Prior to Analysis
April 23rd 2025Researchers leveraged the advantages of thermodesorption, followed by comprehensive two-dimensional gas chromatography coupled to time-of-flight mass spectrometry (GC×GC/TOF-MS), to compare and assess a variety of sampling phases for body odor.



