Dual Flow Chromatography
LCGC Europe
Dual flow chromatography (DFC) separations are performed with back and forth flow for rapid method development, design of experiments (DOE), quality-by-design (QbD), or high-throughput chromatographic purification. Although different than conventional unidirectional flow through chromatography, chromatographic principles still control the separations. Selectivity coefficients and Langmuir adsorption isotherms control the separation chemistry properties of the column and dictate the mobile phase conditions needed to achieve separation. However, the kinetic rates of diffusion and interaction of mobile phase molecules with the stationary phase, column channeling, and other column properties are not germane to the practice of DFC. Chromatographic conditions developed with DFC can be scaled to any size, including laboratory and industrial preparative columns.


Photo Credit: drijimedia/Shutterstock.com

Guenther Bonn1,2 and Douglas T. Gjerde3,1Austrian Drug Screening Institute - ADSI, Innsbruck, Austria, 2Leopold Franzens University of Innsbruck, Innsbruck, Austria 3PhyNexus, Inc., San Jose, California, USA



Dual flow chromatography (DFC) separations are performed with back and forth flow for rapid method development, design of experiments (DOE), quality-by-design (QbD), or high-throughput chromatographic purification. Although different than conventional unidirectional flow through chromatography, chromatographic principles still control the separations. Selectivity coefficients and Langmuir adsorption isotherms control the separation chemistry properties of the column and dictate the mobile phase conditions needed to achieve separation. However, the kinetic rates of diffusion and interaction of mobile phase molecules with the stationary phase, column channeling, and other column properties are not germane to the practice of DFC. Chromatographic conditions developed with DFC can be scaled to any size, including laboratory and industrial preparative columns. One practical use of DFC is in the pharmaceutical industry where multidimensional method development and high-throughput, parallel processing has been used to improve R&D productivity and decrease the time to manufacturing of biopharmaceutical drugs.
Functionalized pipette tips have been available for many years. These pipette tips are used for solid-phase extraction (SPE) and are based on a solid phase contained within a polymer precipitate and located at the end of the tip.
More recently pipette tip columns with conventional chromatographic media packed into beds have been used to improve R&D productivity and capability in academia and industry (1–5). Different stationary phases may be packed into pipette tip columns as required. The packed bed is contained with frits and positioned at the end of a pipette tip. In pipette tip columns, sample and mobile phase enter and exit at the column tip. All liquid flow is bidirectional including sample loading, elution of contaminants, and elution of the desired materials.
In conventional “flow through” chromatography, mobile phase is pumped down through the chromatographic system and column with a pump in a unidirectional flow. Sample is loaded through an injector at the top inlet of the column. Chromatographic separation and elution is a topâdown flow through the column and collection of purified sample may be recovered at the bottom or exit end of the column, opposite to the injection end of the column. The pipette tip chromatographic column may look and operate differently than a conventional unidirectional flow through chromatography column, but chromatographic concepts and principles still control the separation process. This article discusses the principles of pipette tip chromatography operating with bidirectional flow of liquids through the column. We call this dual flow chromatography (DFC).
This article presents the concepts and practical applications of DFC. First, with DFC for pipette tip columns, there are two general aspects to consider. One aspect involves the physical and mechanical properties of the column and how these properties affect the control of flow of fluids through the column. The other aspect involves chromatographic selectivity control of the column to achieve the desired separations. An important feature of DFC makes this most applicable to process development scientists. DFC offers many advantages over traditional methods. DFC is independent of column diameter, flow rates, and amount of resin. DFC is compatible with parallel, multivariate method development. Not only can separation yield be optimized, but also purity, protein activity, nonâagglomeration, and speed can all be optimized in 96-at-a-time experiments. DFC accurately mimics and predicts large diameter traditional column chromatography results for both traditional manufacturing columns and continuous chromatography. With DFC the kinetic rates of diffusion and interaction of samples and eluent with the stationary phase are not important variables to control. This is distinct from conventional, flow through chromatography, especially in miniaturized columns operated in parallel.
Physical and Mechanical Properties of a DFC Pipette Tip Column
The physical and mechanical properties of the pipette tip column are used to control the flow rate and volumes of fluids flowing through the column. In this paper we describe two types of pipette tip columns: packed bed and hollow monolith. One of the key design features of the pipette tip column is the type of frit holding the packing material inside the column. The frits are very thin, typically having a thickness of 60 μm. Interestingly, this is thinner than a typical bed bead. Most agarose-based affinity resin beads have an average diameter of 90 μm. Thus, the thin frit contains virtually no dead volume compared to a column bed volume. For example, a frit containing a 20-µL bed volume has a dead volume of less than 0.25 µL.
The frits of pipette tip columns are hydrophilic. Touching the tip of a column with even a single drop of water will cause water droplets to cover the entire surface of the column (Figure 1). With this property, pipette tip columns have the ability to process very small volumes of liquid. For example, only two to three bed volumes of liquid are needed for elution in a pipette tip column. Thus, a 20-µL bed column requires 40–60 µL of eluent. A 5-µL bed column requires only 10–15 µL of eluent.



This wetting of the frit is a result of hydrogen bonding of water to itself and to the frit. Intra hydrogen bonding of water is often illustrated by the process where small insects can actually walk on top of water. Because the water molecules bind to themselves, the surface of water produces a sort of a surface “skin” or water barrier. Inter hydrogen water bonding and hydrogen bonding of water with the hydrophilic frit will also produces a water “skin” or barrier.
It turns out that this aqueous “skin” is a barrier to air passing through the pipette tip column. The phenomenon is described in Figure 2. Water can pass freely through the frit screen and freely through the column. At the point where there is no more liquid available, a skin barrier is formed at the entrance surface of the frit and flow through the column will stop. Air cannot easily enter the column from either side unless a pressure differential is applied, normally above 2–3 psi. This is called the frit air barrier.


This phenomenon is quite useful in controlling flow through the pipette tip column. Pipette piston position and fluid flow through the column is controlled by pressure and vacuum above the column bed in the chamber of the pipette tip. Software and firmware programming is used to control pipette–syringe piston position to produce predictable vacuum and pressure above the column bed. The piston position and movement does not match the liquid flow, only head pressure dictates flow. The frit itself has essentially zero back pressure and flow through the column only starts after a threshold pressure or vacuum is reached. The flow stops when no liquid is left in front of the frit, and even with a positive pressure or vacuum above the bed air does not enter the column.
An experiment is shown in Figure 1 where a 5-µL bed IMAC affinity column was placed into a well containing 10 µL of liquid. The pipette piston was directed to aspirate, eventually producing a vacuum high enough so that flow starts through the column. The flow of liquid through the column stopped when the liquid was depleted from the well. Air did not enter the column. The process was reversed and repeated several times until the original liquid volume was redeposited back into the well.
Another type of pipette tip column using DFC can be a monolith of polymer with embedded stationary phase material having a hollow central flow path formed at the tip of the column (5). In one example, a divinylbenzene (DVB) monolith with incorporated titanium dioxide (TiO2) and zirconium dioxide (ZrO2) 100-nm nanoparticles was used for selective enrichment of phosphorylated peptides from tryptic digests. The elution volumes were 5 µL with elution volumes as low as 1 µL possible. Other nanoparticle stationary phases may be incorporated into the hollow monolith.
DFC Separation Chemistry Properties
Next we consider DFC separation chemistry properties. Separation selectivity in DFC is controlled by Langmuir adsorption isotherm chemistry, which describes the selectivity of the column for competitive processes. The isotherms show the mobile phase chemical conditions at which sample materials in the mobile phase are adsorbed to the stationary phase column media and the mobile phase conditions at which sample materials are partially dissolved (partially adsorbed the stationary phase) or not adsorbed to the stationary phase and completely in the mobile phase. These sharp isotherm conditions are ideal for performing separations by DFC. Conditions are shown in Figure 3 for two sample materials, one material represented by the dotted line (Material A) and the other represented by the segmented line (Material B). The adsorption and desorption of a molecule for chromatographic media is depicted. The x-axis shows the mobile molecule strength or concentration and the y-axis shows the extent to which a particular material is adsorbed by the resin media.


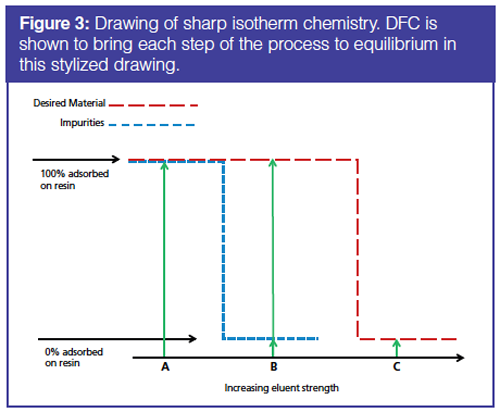
At solvent strength mobile phase Condition A, both the dotted line Material A and the segmented line Material B are adsorbed completely on to the column. This is the capture step. As the mobile phase strength is increased to mobile phase Condition B, the segmented line material completely desorbs or elutes from the resin, but the dotted line material is still completely on (adsorbed to) the resin. This is the wash step where nonspecific bound or nontarget sample materials (dotted line Material A) are removed from the column, but the targeted sample (Material B) remains adsorbed to the stationary phase. Finally, at mobile phase eluent Condition C, the elution step, the dotted line of the desired sample (Material B) completely desorbs from the resin and is recovered. So with sharp isotherms and with step gradients, separation conditions can be achieved. If selectivity permits (selectivity coefficients are sufficiently different), several step gradient elutions may be employed to recover distinct fractions of components (5).
The interaction of biomolecules with most chromatographic phases can be characterized by sharp isotherm “on-off” chemistry. Under any set of buffer conditions, the biomolecule is either adsorbed to the chromatography material or desorbed from the material. This appears to be true for ion-exchange chromatography, ion-pairing chromatography, affinity, hydrophobic interaction chromatography, reversed phase chromatography, hydrophilic interaction chromatography, and normal phase chromatography.
Sharp isotherms are ideal for performing separations (Figure 3). Solvent strength Condition A shows the buffer conditions at which a column is loaded. Both the desired protein drug (red segmented line) and impurities (blue segmented line) are adsorbed on to the column. As the separation method proceeds the eluent strength is increased to eluent Condition B. Impurities ideally completely desorb from the resin. However, the red material, the material of interest, is still completely bound to the resin. Depending on how tightly bound to the resin, the stringency of the wash may be increased to remove as many impurities as possible without removing the desired material and without harming or denaturing the desired material. Finally, at eluent Condition C, the desired material completely desorbs from the resin and can be recovered. So with sharp isotherms, separation methods can be developed.
In practice, the isotherms are not actually as sharp as depicted in Figure 3. To obtain sharp isotherms, a number of parameters are tested and controlled. Wash conditions are carefully selected so that desired product remains on the column while undesired materials are removed and washed away. If the target biomolecule to be recovered is a relatively small molecule, the binding is usually relatively high. Tagged recombinant proteins or protein fragments generally have a single point of attachment to the affinity resin. An example is HIS-tagged proteins or fragments that have affinity for IMAC resins. Smaller molecules that have a single point of attachment bind tighter to resin than a larger molecule as a result of steric hindrance or other factors. On the other hand, if the number of binding sites increases with size, then the selectivity may increase with size, again depending on steric hindrance and other factors. Stringent conditions can be chosen for washing without fear of removing the target biomolecule. With stringent washing, nonspecific materials are efficiently removed prior to elution of the pure target material. However, strong elution conditions may be needed for complete recovery.
If the sample biomolecule is relatively large, it may not bind as tightly to the stationary phase. The stringency of the wash is normally reduced to prevent loss of the target material. Conditions for washing are carefully chosen to remove as much nonspecific material as possible while still retaining the target material. However, elution is relatively easy and high recoveries from this step are possible, provided sample material was not lost in the wash step. Thus, conditions for capture, wash, and elution are developed for each case to optimize the mass recovery, concentration, and purity of the biomolecule. In addition, the conditions are chosen to keep biomolecules active and not denatured, which will be described later.
The ability to achieve complete isotherm equilibrium of stationary phase and mobile phase interaction can be difficult to achieve, but parallel processing of pipette tip columns to study multiple variables simultaneously provides a convenient and practical solution for achieving this. Biomolecules adsorb slowly with chromatographic stationary phase columns. The kinetics of diffusion of molecules to and into the stationary phase bead and to functional groups is slow. The kinetics of the actual binding interactions is slow. The problem is exacerbated with small columns, especially spin columns and plates where flow through is extremely rapid. The time of interaction of the sample with the column is difficult to control and may be insufficient. Linear velocity of mobile phases travelling through the column increases as the column volume and (in particular) the column diameter is decreased.
Increasing the time of interaction will compensate for slow reaction kinetics. The time of column interaction is easily controlled with DFC simply by controlling the number of back and forth flow cycles until equilibrium interaction of sample and column are achieved. This is shown by Figure 4 where 5-, 10-, 40-, and 160-µL pipette tip columns of Protein A were used to capture an IgG. The smallest column having the fastest linear velocity of fluid flow takes the longest time to achieve complete interaction. In any case, within 3 min of back and forth flow interaction, the capture is complete, regardless of the bed size.



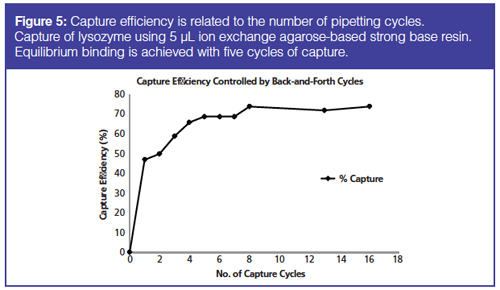
In Figure 5 the capture of a protein on a cation exchange column is plotted as a function of the number of back and forth cycles. Several cycles are needed for capturing lysozyme protein. The capture is at equilibrium with five cycles and increasing the interaction further only gave slight improvement.


Stationary phase and molecule interaction is driven to completion with DFC regardless of the kinetic rate constants. Because the interactions are at equilibrium, DFC separations are independent of:
- Linear and absolute fluid flow rate through the column,
- Column bed diameter, length, and geometry,
- Packing uniformity or column channelling,
- Column dead volumes, and
- Amount of stationary phase in the column.
With sufficient time of interaction (achieved through dual directional flow) equilibrium interaction is achieved regardless of these parameters. Channelling along the sides or through channels of the packed bed are not factors for how well a pipette tip column performs. Unlike flow through columns, the bed packing does not have to be uniform. The interaction is only dependent on column stationary phase and mobile phase chemistry making DFC suitable for miniaturized-, parallel-, and scalable chromatography.
Equilibrium reaction is achieved even with small volumes of step gradient buffers. For 5-µL bed columns, the step gradient volume may be as small at 10 µL. In this way, fine separation control may be achieved through controllable and repeatable concentrations of the mobile phase steps that are applied to the stationary phase.
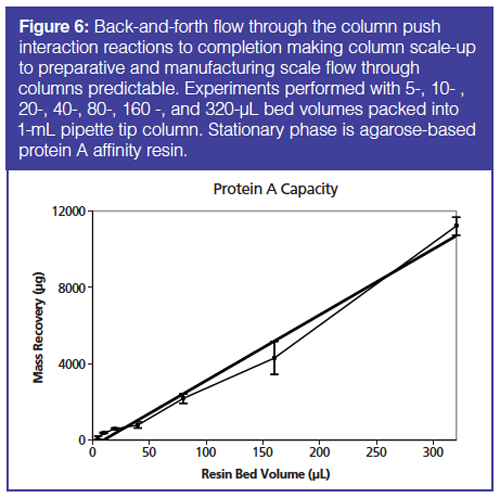
Scalability is shown in Figure 6 where there is a linear relationship between pipette tip resin bed volume and recovery of protein. This plot shows recovery of an IgG protein from 5-, 10-, 20-, 40-, 80-, 160-, and 320-µL bed volume columns. The column capacity was found to be as stated by the resin manufacturer’s specifications.


The separation conditions developed with these small columns can be applied to all column bed sizes, including laboratory preparative columns and large-scale manufacturing. DFC columns mimic and predict the performance of preparative and manufacturing columns. This is because preparative columns typically have a large column diameter. This in turn makes the linear velocity of fluid flowing through the column very slow. Contact time is long and equilibrium interaction is achieved, just as in pipette tip columns.
Wenger and coworkers applied this principle as described in reference 6. Work was performed to determine the 1âm diameter column conditions needed for manufacturing HPV, a vaccine to prevent cervical cancer. Conditions for manufacturing columns were developed with 80-µL bed pipette tip columns reducing the time to production for the drug by several months.
Tip Concentrating Effect
With DFC, the resin in the small column bed is used so efficiently that the sample becomes very concentrated when it is collected. Very high concentrations of recovered protein can be achieved, in some cases up to 10 mg/mL (7). To demonstrate, a comparison of a Protein A spin column and a 20-µL bed Protein A pipette tip column was performed. Both columns use capture, wash, and elution steps. However, the spin column is a unidirectional flow column with larger dead volumes and the pipette tip column uses back and forth flow. The results are shown in Figure 7. The sample is an antibody that was spiked into an E. coli cell lysate. Identical samples were applied to three Pierce spin columns and purified and the recovered samples separated on three lanes of the gel. No protein bands can be seen indicating that either the antibody was not recovered or the recovered antibody is very dilute compared to the original sample. Next, the sample was applied to three 20-µL bed pipette tip columns. The gel bands show that the recovered protein concentration is much higher than protein recovered from the spin column. The concentration of proteins recovered from pipette tip columns are also much higher than the starting sample.

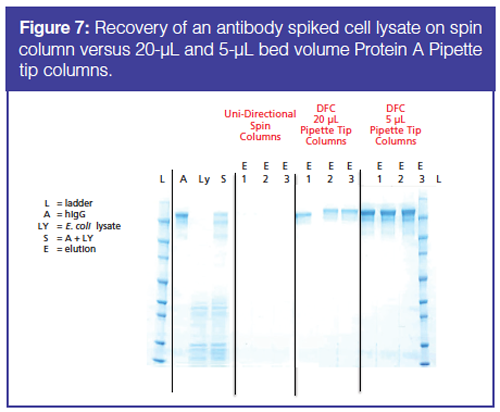
Efficient use of the pipette tip column to collect material is termed the tip concentrating effect. The tip concentrating effect can be illustrated further by extending the experiments and decreasing the pipette tip column size to 5 µL from 20 µL. The results (Figure 7) show that as the column is decreased to the smaller 5-µL bed column significantly higher concentrations of protein are recovered. Although counterintuitive, the smaller bed column produces higher concentrations of protein. As the resin bed is decreased, the resin bed can be loaded more effectively. “Loaded” columns provide capacity data because the columns are brought to equilibrium. These data can be used to calculate the projected yield of manufacturing-scale single columns and continuous chromatography systems. At the same time, the dilution effects of removing the protein from the column are limited because of the very low dead volume of the pipette tip column.
Concentrations and recoveries of proteins from the experiment are shown in Table 1. The spin columns produced larger volumes of purified protein but the concentrations are extremely low. Proteins would have to be reconcentrated to make this useable for any type of downstream processing. The 20-µL bed pipette tip column produces useable, higher concentrations of proteins. Finally, the smaller 5-µL bed pipette tip column produces even higher concentrations of protein, although at a cost of lower mass and percent recovery.


To summarize, the smaller column will produce the highest concentration of protein provided the larger column is not overloaded. Because equilibrium of capture is achieved, the researcher may match the bed size to sample concentration and volume to load up the affinity resin bed and achieve high recovered concentrations.
Parallel Operation and Automation
There are generally two reasons for parallel operation of pipette tip columns. One is to determine the chromatographic conditions to optimize purity and recovery of the product. This can be accomplished easily by using a 12-column parallel robotic pipette head operating with a 96-well plate. Various sample loading conditions, column wash conditions, and elution conditions can be operated along the 12-well plate axis and eight conditions as the robotic head moves along the eight-well plate axis (or 16 conditions if two plates are used in series). This is illustrated in an example experiment where the variables include 12 types of resins for each row (repeated eight times in a box of 96 columns) with four different buffer capture conditions (varying the pH). Each resin type may capture the sample in duplicate with a particular buffer. In this example, there may be two different wash conditions (changing the ionic conditions or pH) and three different elution conditions performed in steps changing the ionic conditions or the pH. In total, 288 conditions can be tested in one set of experiments.
Interestingly, high protein recovery and high protein activity are not necessarily correlated, showing the need for this type of method development. Experiments such as these can be performed in a few hours compared to flow through column method development that may take many days or weeks. In addition, it is quite easy to study two or more variables, such as the effect of pH and salt concentration, in the same set of experiments. The complex interactions of sample and contaminants with the resin can be studied to optimum yield.


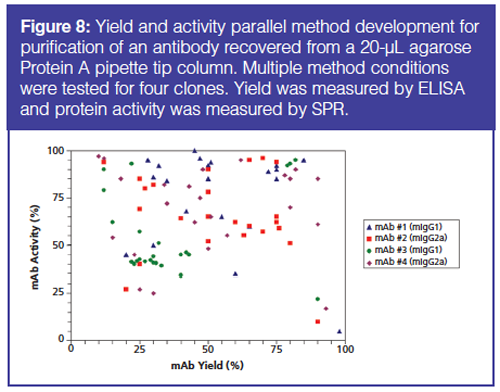
In protein purification, not only is yield important but also the recovered protein must remain active and not agglomerated. Figure 8 shows the results of an experiment of parallel method development for purification of an antibody on one type of Protein A column. Different conditions of capture, wash, and elute (eight each) were tested for each of four clones. The recovered proteins were screened for yield (ELISA) and retention of activity (SPR). The results show that high recovery and high (retained) activity are not correlated and both goals should be considered when developing separation conditions for proteins. It is difficult to consider these goals without parallel purification.
When separation conditions are established, parallel operation of columns can be used to separate up to 96 samples at a time. Some robotic liquid handlers may be loaded with 10 to 30 plates of samples for a single operational run. Table 2 shows the reproducibility of recovery of a protein operating with DFC columns in parallel.

Conclusion
Dual flow chromatography (DFC) with sample and mobile phase travelling in both column directions represents a new way to study and develop separations. DFC with pipette tip columns is suitable for miniaturized, parallel, and automated operation. Separation conditions developed for small columns are scalable. Conditions developed on columns as small as 5-µL bed size can be transferred to industrial manufacturing columns.
DFC works with any column-analyte
chemistry where the Langmuir adsorption isotherms are sharp. Biomolecules have sharp isotherms for most different types of chromatographic phases including mixed mode resins. Although kinetic rates of diffusion and interaction can be slow for the small bed columns, with DFC interaction of samples with stationary phase and mobile phase are driven to equilibrium. Equilibrium reaction is achieved even with small volumes of step gradient buffers. For 5-µL bed columns, the step gradient volume may be as small at 10 µL.
Parallel DFC chromatography can be used for method development including multivariable condition determination, which is difficult to perform in unidirectional chromatography. DFC is useful in the competitive (bio)pharmaceutical industry where speed is critical for method development to determine separation conditions and for high-throughput parallel sample processing. DFC is also useful for sample preparation for analytical purposes, especially where high through put sample processing is needed and only limited sample is available.
References
- M. Ramakrishnan, F. Alves De Melo, B.M. Kinsey, J.E. Ladbury, and T.R. Kosten, PLoS ONE7(7), e40518 (2012).
- B.D. Prater, K.R. Anumula, and J.T. Hutchins, Analytical Biochemistry369(2), 202–209 (2007).
- R. Hopkins, D. Esposito, and W. Gillette, J. Struct. Biol.172(1), 14–20 (2010).
- M. Olajos, A. Szekrenyes, P. Hajos, D. Gjerde, and A. Guttman, Analytical and Bioanalytical Chemistry397(6), 2401–2407 (2010).
- M. Rainer, H. Sonderegger, R. Bakry, C.W. Huck, S. Morandell, L.A. Huber, D.T. Gjerde, and G.K. Bonn, Proteomics8, 4593–4602 (2008).
- M.D. Wenger, P. DePhillips, C.E. Price and D.G. Bracewell, Biotechnol. Appl. Biochem. 47, 131–139 (2007).
- D.T. Gjerde, U.S. patent 8,143,071, 21 December 2007.
Guenther Bonn is Founder and Director of the Austrian Drug Screening Institute (ADSI) and Executive Director of the Institute of Analytical Chemistry and Radiochemistry at the University of Innsbruck. Bonn has published over 350 papers, more than 30 patents, has co-authored two books, and possesses global expertise in areas related to the fundamental chemistries and integration processes involved in biomolecular separations, particularly with respect to macromolecules such as nucleic acids and proteins as well as phytoanalysis. Professor Bonn has longtime industry connections and leads numerous scientific projects together with industrial companies in Austria and abroad. He serves as a member of the board of Bionorica SE. Professor Bonn has received many national and international awards, including the Halasz Award, the Csaba Horváth Memorial Award, and the Honorary Ring of the Austrian Academy of Sciences.
Douglas T. Gjerde received his Ph.D. in analytical chemistry from Iowa State University under Professor Jim Fritz. After working in large and small companies for a number of years, Gjerde founded Sarasep in 1990 for the production and commercialization of polymeric separation technologies. Profits derived from this business funded the development and commercial introduction of the first commercial high performance liquid chromatography separations of DNA. In July 1997, Gjerde cofounded Transgenomic, Inc. helping bring the company public in July 2000. Gjerde founded PhyNexus in 2003 bringing the first products to market Gjerde has authored and coauthored numerous articles, six books, and holds over 80 patents in separation science.








; An Interview with Fabrice Gritti")
: An Interview With Fabrice Gritti")


Determining Enhanced Sensitivity to Odors due to Anxiety-Associated Chemosignals with GC
May 8th 2025Based on their hypothesis that smelling anxiety chemosignals can, like visual anxiety induction, lead to an increase in odor sensitivity, a joint study between the University of Erlangen-Nuremberg (Erlangen, Germany) and the Fraunhofer Institute for Process Engineering and Packaging (Freising, Germany) combined behavioral experiments, odor profile analysis by a trained panel, and instrumental analysis of odorants (gas chromatography-olfactometry) and volatiles (gas chromatography-mass spectrometry).


Investigating 3D-Printable Stationary Phases in Liquid Chromatography
May 7th 20253D printing technology has potential in chromatography, but a major challenge is developing materials with both high porosity and robust mechanical properties. Recently, scientists compared the separation performances of eight different 3D printable stationary phases.
. | Image Credit: © Joan Vadell - stock.adobe.com")
Detecting Hyper-Fast Chromatographic Peaks Using Ion Mobility Spectrometry
May 6th 2025Ion mobility spectrometers can detect trace compounds quickly, though they can face various issues with detecting certain peaks. University of Hannover scientists created a new system for resolving hyper-fast gas chromatography (GC) peaks.


