Direct Enantiomer-Selective Mass Spectrometry of Chiral Mixtures by Mass-Selected Photoelectron Circular Dichroism
Special Issues
Simultaneous, enantiomer-specific identification of chiral molecules in multicomponent mixtures is extremely challenging. With mass-selected photoelectron circular dichroism (MS-PECD) using an electron–ion coincidence imaging spectrometer, a compound can be identified as chiral without the need for any prior enantiomeric separation or enantiomer-selective complexation.
Simultaneous, enantiomer-specific identification of chiral molecules in multicomponent mixtures is extremely challenging. Many established techniques for single-component analysis fail to provide selectivity in multicomponent mixtures and lack sensitivity for dilute samples. Mass spectrometry is chirally blind and cannot directly distinguish the two enantiomers of chiral molecules. Here we discuss how enantiomers may be differentiated by mass-selected photoelectron circular dichroism (MS-PECD) using an electron-ion coincidence imaging spectrometer. Following an ionizing circular polarized laser pulse, ions and electrons are detected in coincidence on their respective time- and position-sensitive detectors. The MS-PECD asymmetry measured on electrons tagged by the mass of their corresponding parent ions directly reveals that the compound with identified mass is chiral without the need for any prior enantiomeric separation or enantiomer-selective complexation. MS-PECD enables direct enantiomeric excess measurement of multicomponent chiral samples in a benchtop mass spectrometer.
During the last few decades mass spectrometry (MS) has become the most important analytical technique in research and development (1,2). In 2015 the global MS market revenue was about $3 billion (3). Within the pharmaceutical industry MS has become the workhorse technique to improve drug safety and reduce costs associated with drug discovery and development processes. Also within clinical applications such as biological tissue research (4,5) and diagnostic testing in hospitals (6), MS techniques are rapidly proliferating.
MS is, however, inherently chirally blind and cannot directly distinguish the two enantiomers of chiral molecules. Nevertheless, within the bio-ecosphere a preferred chiral form of very many molecules is dictated by the chirally sensitive binding efficacy to bioreceptors and their biological function. Analytical techniques that can distinguish the enantiomers of chiral molecules are, therefore, desirable and indeed have become extremely important in pharmaceuticals (7), foods and fragrances (8), and agrochemicals (9). Chiral molecules obviously create a new challenge for MS approaches.
The common ionization techniques used in MS (electron impact, matrix-assisted laser desorption–ionization [MALDI], electrospray ionization [ESI]) have no chiral selectivity. Therefore, some other form of chiral interaction, like enantiomer-selective binding (10,11) or enantiomer-selective separation (12), must be introduced before MS techniques can be used for chiral detection and mass analysis.
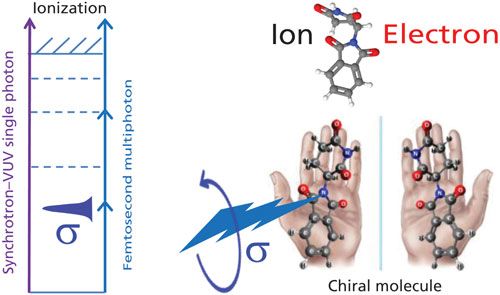
Ionization of molecules can, of course, be readily achieved by electromagnetic radiation (Figure 1). In general one needs about 8–12 eV to ionize a molecule, so either high-energy vacuum ultraviolet (VUV) photons, from a source such as a synchrotron, or multiple (less energetic) photons, generated by an intense (ultrafast) laser, are used. In the ionization process both an ion and an electron are produced, and determination of the mass of the ions alone is usually the total information MS tools provide. However, this article describes how the electron, which normally goes undetected, can provide a wealth of information about the molecule. In particular, we discuss the enantiomer selectivity that the electron can provide upon ionization of a chiral molecule.


Figure 1: Schematics of the formation of an ion and an electron in the photoionization of a chiral molecule by VUV single-photon or laser-based multiphoton excitation.
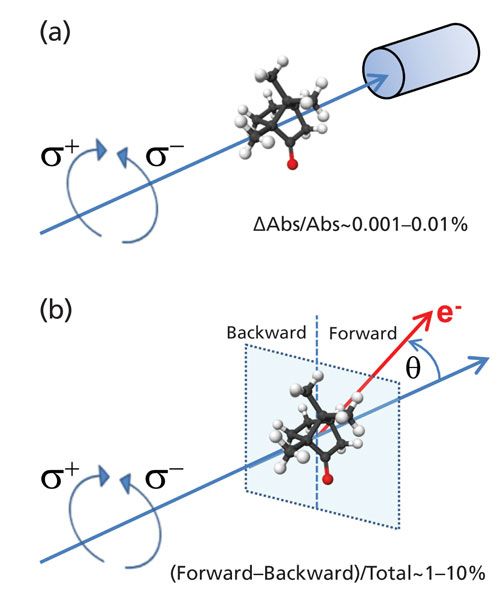
As a basic principle, one needs a chiral reference system to detect enantiomers of chiral molecules. A fully generic chiral reference is provided by circularly polarized light, readily prepared and characterized by well-established optical techniques. Chiroptical methods, such as electronic and vibrational circular dichroism (CD, VCD) and vibrational optical activity (VOA) exploit the often small chiral asymmetries in light–matter interaction (see Figure 2), but are widely used techniques for the detection and structural determination of solvated chiral molecules (13). Electronic and vibrational CD and VOA rely on an interference effect between electric dipole and magnetic dipole transitions, and because of the generally limited transition strength of magnetic dipole transitions the CD signal sensitivity is generally rather weak, typically only about 0.001–0.01% of the total absorption strength. In certain cases it was recently shown that the CD effect can be enhanced significantly by influencing the electronic structure of the solvated enantiomers by adding metal ions to the solution (14). The measurement of CD and ion detection in pulsed UV–vis laser excitation was pioneered by Boesl (15) and Weitzel (16). Another enantiomer-selective spectroscopic method was recently developed that relies on dipole selective microwave spectroscopy using newly developed chirped RF-generators (17,18). In this latter technique, the phase-sensitive detection of a microwave signal after selective excitation of coupled rotational energy levels provides enantiomer sensitivity.

Figure 2: Concept of (a) circular dichroism and of (b) photoelectron circular dichroism (PECD) (44,47).
Mass-Selected Photoelectron Circular Dichroism
Here we present a novel technique that is directly enantiomer selective and is coined mass-selected photoelectron circular dichroism (MS-PECD). PECD is a phenomenon that can be observed when a chiral molecule is photoionized by circular polarized light and the electron that is formed in the ionization is detected, see Figure 2. In 1976 Ritchie (19) published a theoretical paper showing for the first time that when you ionize a chiral molecule, the angular distribution of the ejected electron will show a profound forward–backward asymmetry in the number of electrons ejected along the propagation direction of the ionizing radiation (forward) or in the opposite direction (backward). For instance, Ritchie predicted in a calculation of a chiral model potential that when you ionize the R-enantiomer with circular polarized light you will see up to 10% more electrons ejected at 0° with the propagation direction of the light relative to the amount of electrons ejected at 180°. When you reverse the helicity of the light from left-circular-polarized (LCP) to right-circular-polarized (RCP) the electron asymmetry would switch sign, so 10% more electrons would be ejected in the backward direction. Or if you would switch the chiral molecule from R- to S-enantiomer, and would keep the same helicity of the light, the electron asymmetry would also switch sign, and 10% more electrons would get ejected in the backward direction. Ionization of a nonchiral molecule would never show such forward–backward asymmetry in electron intensity. In early 2000, the first realistic PECD calculations of real molecules like alanine were published (20) and shortly afterwards the first experimental demonstrations of the PECD phenomenon appeared using synchrotron radiation (21,22).
As illustrated in Figure 1, it is also possible to photoionize a molecule via multiphoton excitation. Using an ultrafast laser it was demonstrated already in 1991 that it is very easy to ionize molecules and do mass spectrometry detection of gaseous samples to study their photodynamics (23). Furthermore, around 2000, the first femtosecond laser experiments demonstrated how it is possible to study the spectroscopic ultrafast photodynamics of molecules by ionization and simultaneous detection of both ions and electrons from the same event using coincidence imaging particle detectors (24,25). This requires fast time-response detectors (26–29) to capture the temporal correlation between ion and electron. With this correlation the experimentalist can identify the electron–ion pairs originating from a single molecular ionization event. Adding spatial sensitivity by using fast imaging position-sensitive detectors enables the recording of angular distributions of such correlated pairs in coincidence detection mode.
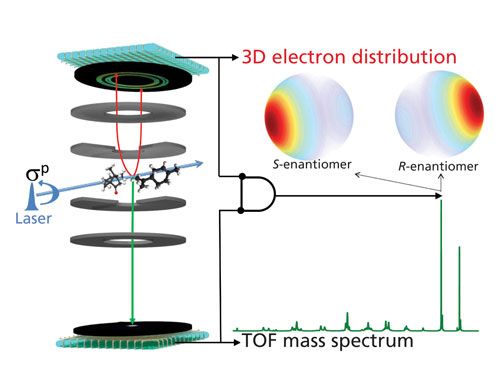
In 2011 and 2012, the Amsterdam group used a homebuilt electron–ion coincidence setup (27,29) to employ PECD with ultrafast lasers and they reported the first demonstration directly measuring the enantiomers of camphor with mass-selectivity (30,31). This technique was named MS-PECD (Figure 3). A circular polarized ultrafast laser near 400 nm ionizes the camphor molecules in a gaseous expansion. For every laser shot, one observes whether an ionization event happens, and the electron is extracted in a velocity map imaging (VMI) (32) charged particle lens to a time- and position-sensitive detector. Because of the light mass of the electron the arrival time is usually within tens of nanoseconds of the laser pulse. In the opposite direction, using either time-of-flight (TOF) detection or TOF combined with VMI imaging detection, the correlated ion is detected after flight times of several microseconds. The electron and ion events are time-stamped by the time-sensitive detectors and the electron and ion can be correlated in the analysis (Figure 3). The delayed signal from the ion detector directly provides the mass of the detected species as in normal TOF-MS detectors (33). On the electron imaging detector the full three-dimensional (3D) momentum distribution of the electron is obtained. Depending on the wavelength of the laser and the final internal state of the cation, the electron can be ejected with a distribution of kinetic energies. The higher the photoelectron energy the larger the distance the electron can travel perpendicular to the extraction direction of the electron time of flight, and so the larger the 3D sphere of electron momentum imaged at the detector. Furthermore, the angular anisotropy on the electron detector gives the amount of electrons ejected in the forward (θ < 90°) hemisphere or in the backward (θ > 90°) hemisphere. In fact, with multiphoton excitation a structured oscillating angular pattern may be formed that provides much insight on the energy levels and transition dipoles involved in the ionization process (34,35).

Figure 3: Concept of mass-selected photoelectron circular dichroism. A mixture of chiral molecules is photoionized by a circular polarized laser pulse. The three-dimensional momentum distribution of the electron is imaged on a time- and position-sensitive detector, the time of flight of the ion is measured on a second time-sensitive detector. The correlation of the two events provides direct mass-tagged enantiomer selective data on chiral molecules in complex mixtures. Adapted with permission from reference 45.
The first experiments in Amsterdam using MS-PECD were done on camphor, a molecule that was independently studied by the Kassel group with laser excitation, but using a different detector that is only able to detect the electron distribution (36). In 2013, a full experimental account on multiphoton MS-PECD was reported by the Amsterdam team in collaboration with the Nottingham group of Powis providing also a theoretical analysis and interpretation of the magnitude of the multiphoton PECD signal (35). The enantiomer sensitivity of camphor was about 8% for laser excitation at 400 nm, about a factor of 2 larger than the sensitivity measured by synchrotron single photon VUV excitation (37,38) at similar electron kinetic energy.
After publication of the first experimental results of mass-selected PECD on camphor, other chiral molecules like methyloxirane and limonene were studied in Amsterdam (39–41). The Kassel group presented more-detailed PECD studies on camphor and fenchone with electron detection only (42,43). Furthermore, the synchrotron group of Nahon at Soleil with Powis has provided a multitude of PECD studies during the last decade using single VUV photon ionization demonstrating the sensitivity of PECD to vibrational effects and cluster dynamics (44).
Direct MS Measurement of Enantiomeric Excess in Mixtures
The tremendous advantage of MS-PECD is that for an arbitrary mixture of chiral molecules the enantiomeric excess can be determined directly. Here we want to highlight the recent experiment from the Amsterdam/Nottingham team (45) that clearly demonstrates the analytical MS potential of mass-selected PECD. By preparing various mixtures of the two enantiomers of the chiral molecules camphor and limonene, coexpanding in a gaseous flow of neon, it was possible to measure the enantiomeric excess directly in a diluted mixture of molecules without either prior enantiomeric separation, such as by supercritical fluid chromatography (12), or complexation (10). The gas flow of mixture molecules was crossed by an ultrafast laser and the ion and electron were detected in coincidence as shown in Figure 3. Some of the results are summarized here, focusing on the information obtained from the electron distribution. Because the masses of an enantiomer pair are identical, the TOF mass spectra show no differences between the different mixtures. A representative example from one of the mixtures is shown in the lower part of Figure 3, clearly showing the two parent masses of limonene at mass 136 and camphor at mass 152. One can even observe the small peaks at masses 137 and 153 corresponding to the parent masses with natural abundance of the 13C isotope. Several other minor peaks are observed due to fragmentation of the parent ions after additional photon absorption. In this experiment the laser power and focusing was set to minimize parent fragmentation and provide soft ionization. However, by increasing the fluence of the laser pulse one can increase the fragmentation of the parent ion (for example, see Figure 5 in reference 35). The typical TOF mass resolution (M/ΔM) of the ion detector is about 4500 (46), when optimizing the ion optics for masses in the region 100–200 amu and utilizing only a simple linear TOF tube with a length of about 30 cm. The mass resolution benefits from ultrafast laser ionization with a short laser pulse duration (~150 fs), the perpendicular ion extraction of a small source region of some 50–100 µm in diameter along the TOF axis (27,29), and a fast timing detector with small-pore microchannel plate detectors with fast read-out electronics (27). For a recent discussion on TOF-MS and the experimental issues that determine resolution, see the recent review by Boesl (33).
We now focus on the information obtained from the electron. In the top panel of Figure 4, the total kinetic energy spectrum is shown for all photoelectrons when the mixtures are ionized with right-circular-polarized laser radiation at around 392 nm. This means that the kinetic energy was extracted from the 3D data (t,x,y) of the electron detector without any correlation of electron events with a particular mass ion. The top panel shows two curves, which are similar within experimental accuracy, measured for two different mixtures, one mixture having R-enantiomers of both limonene and camphor, mix-1 = (R136/R152) and a mixture having the R-enantiomer of limonene and the S-enantiomer of camphor, mix-2 = (R136/S152). As expected, the total photoelectron energy spectrum does not change when you change the camphor enantiomer. It doesn’t matter which enantiomer is ionized, the energy levels of the enantiomers are the same, the extremely small energy shifts due to parity violation cannot be observed, and the photoelectron kinetic energy spectrum is the same. Similar curves, not shown here, were measured when photoionizing with left-circular-polarized laser light.
The spectra in the top panel are rather broad and showing some minor indication of underlying structure. The middle panel of Figure 4 shows the energy spectrum obtained when only the electrons that were measured in coincidence with ions of mass 136 (limonene) were selected out of all the coincidence events, and in the bottom panel of Figure 4 for electrons measured in coincidence with ions of mass 152 (camphor). Again the two curves in both panels are for the two mixtures (R136/R152) and (R136/S152). Now the electron energy spectra of coincident events are clearly different. The electron spectrum measured for limonene ions (middle panel) shows a peak around 0.38 eV and a smaller bump around 1.0 eV. The electron spectrum for camphor ions (bottom panel) is narrower in structure and peaks around 0.6 eV. With the known ionization energy of camphor one can directly deduce that the ionization process involves three 392-nm photons. The electron spectrum of limonene provides more-complex spectroscopic information, and further details are reported elsewhere (40,41). As expected, the photoelectron spectra for the mass-tagged electrons also shows no difference for the two mixtures (R136/R152) and (R136/S152).

Figure 4: Three photoelectron energy panels (left) obtained with right-circular-polarized light for a mixture of R-limonene/R-camphor (red curves) and R-limonene/S-camphor (blue curves). The top panel is the kinetic energy distribution of all electrons independent of the correlated ion, the middle panel the same for mass-tagged electrons in correlation with limonene (m = 136 amu), the bottom panel for mass-tagged electrons in correlation with camphor (m = 152 amu). The right side of the figure shows the measured PECD asymmetry G (in %) for three different mixtures while detecting all electrons (top row), limonene tagged electrons (middle row), and camphor tagged electrons (bottom row). The label (R136/R152) is for mix-1 containing the R-enantiomer of limonene and the R-enantiomer of camphor, the label (R136/S152) is for mix-2 containing the R-enantiomer of limonene and the S-enantiomer of camphor, the label (S136/Rs152) is for mix-3 containing the S-enantiomer of limonene and a 3:1 composition of R:S-enantiomers of camphor. Adapted with permission from reference 45.
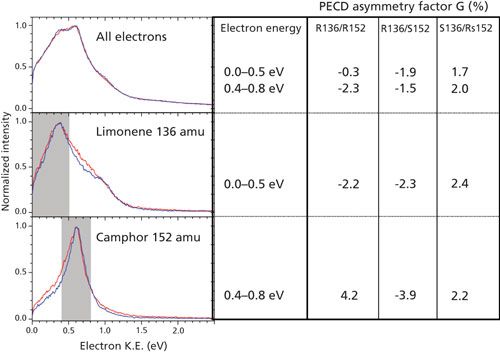
Subsequently, we provide the PECD asymmetry factor G (35) that is determined by looking at all the electrons emitted in the forward hemisphere versus the electrons emitted in the backward hemisphere. To cancel all experimental spatial inhomogeneity of the detection we do measurements for RCP and LCP light. The two data sets are subsequently normalized and subtracted in such a way that one is only sensitive to the asymmetry due to the PECD effect (35) and all experimental factors are canceled out to first approximation. The right side of Figure 4 shows this enantiomeric asymmetry factor G, for three mixtures mix-1 = (R136/R152), mix-2 = (R136/S152), and mix-3 = (S136/75%-R152/25%-S152). The third mixture (mix-3) switches from R- to S-limonene (compared to mix-1 and mix-2) and also contains 3:1 R:S camphor corresponding to an enantiomeric excess (ee) of 50%.
The PECD asymmetry G for tagged electrons from limonene, with energy in the grey region of 0–0.5 eV, is around 2.3%. G(136) switches sign nicely between mixtures 1,2 (R136) and mixture 3 (S136), the sign being negative for R-limonene (and RCP) and positive for S-limonene. For camphor (ionized around 392 nm), the asymmetry G(152) = 4.2% and switches also nicely in sign between mix-1 and mix-2. Furthermore, because the measured asymmetry G for electrons tagged with mass 152 is directly linear proportional to the enantiomeric excess of camphor, knowing the G-value for both the R- and S-enantiomers of camphor permits the observed value of Gmix-3(152) = 2.2% to be directly converted to ee(152) = 50% (45).
This measurement shows the power of mass-selected PECD for direct measurement of enantiomeric excess in diluted (1%) multicomponent mixtures of chiral molecules in an inert carrier gas, without any prior enantiomeric separation or complexation. A calibration has to be made only once to obtain the G-factors for pure R- or S-enantiomers of the components. Following a one-time calibration against pure components, subsequent measurements of chiral components gives direct and separate quantitative measurement of enantiomeric excess for each chiral mass that can be tagged to the electrons observed in the electron-ion coincidence measurement.
Future Prospects
Around 2000, the first experimental demonstrations of PECD using synchrotron ionization were performed (21,22) and up to around 2011 all experiments were done at a few synchrotrons capable of delivering circular polarized radiation and with advanced imaging detectors (44). With the extension of PECD to benchtop laser-based experiments starting around 2011, the PECD field is growing rapidly with the prospects of a variety of analytical and research applications on chiral molecules (47). It is important for further growth of the PECD technique and applications that the experimental efforts are accompanied with high-level theoretical calculations. After the development of reliable one photon ionization scattering calculations (48), recent theoretical calculations were also reported for multiphoton resonant excitation (35,49) and strong-field PECD modelling (50,51). In 2015, other laser-based techniques for the production of energetic VUV radiation, such as circular polarized high-harmonic-generation (52,53), have been reported that were used in single-photon PECD measurements. These benchtop laser experiments have demonstrated that VUV generation by lasers is also a viable path for sensitive spectroscopic studies of chiral molecules employing PECD. It was further shown in 2016 that PECD is a very general phenomenon that can be observed in single-photon, multiphoton, and even strong field ionization of chiral molecules (54). Also, femtosecond time-resolved PECD studies were published recently, demonstrating the sensitivity of PECD to vibronic coupling and nonadiabatic electronic coupling (55,56) in chiral molecules.
Furthermore, two recent studies in 2016 using both synchrotron radiation (44) and multiphoton laser excitation (57) have shown that the accuracy of enantiomeric excess measurements in single component PECD experiments have now reached the 1% level. Because the asymmetry G is obtained from a simple counting of forward and backward electrons, the error in the measurement of G is determined by Poisson statistics (45). It can be shown that for G-factors in the region of 1–10% the absolute accuracy is approximately given by ΔG ≈ 2/Ne½, where Ne is the number of electrons counted. This means that to obtain a 1% relative measurement error (ΔG/G = 1%), when G = 5%, Ne should be larger than 4/(0.01*0.05)2 ≈ 16 million electrons. It is clear that a large average count rate will enable short measurement times, and when the count rate increases to about 105/s a couple of minutes of counting is sufficient to reach 1% accuracy in enantiomeric excess measurements. These benchtop analytical experiments warrant high-repetition rate laser light sources, and such developments are rapidly progressing within the laser photonics field (58).
High-repetition-rate laser experiments will open the mass-selected PECD technique to benchtop analytical applications of direct and rapid MS detection of chirality in multicomponent mixtures of chiral molecules and the accurate quantitative enantiomeric excess measurements in complex chiral samples (45,47).
Acknowledgments
This research has been financially supported by the council for Chemical Sciences of the Netherlands Organization for Scientific Research (NWO–CW/VICI,BAZIS) and by EPSRC (UK). The authors acknowledge support by the European Union through the Integrated Infrastructure Initiative LaserLabEurope and for synchrotron access via European Community-Research Infrastructure Action and European Community’s Seventh Framework Program. We also acknowledge support through the EU Marie-Curie Initial Training Networks ICONIC and ASPIRE. The authors gratefully acknowledge Drs. M. M. Rafiee Fanood, C. S. Lehmann, N. B. Ram, W.G. Roeterdink, and A. Vredenborg for all the collaborative research in Amsterdam and the excellent technical support by Mr. R. Kortekaas. I.P. greatly acknowledges the invaluable long standing collaboration with Drs. L. Nahon and G. Garcia at Synchrotron Soleil.
References
- F.W. McLafferty, Ann. Rev. Anal. Chem.4, 1–22 (2011).
- L. Tokes, Mass Spec.Rev., DOI: 10.1002/mas.21499 (2016).
- Frost and Sullivan, Analysis of the Global Mass Spectrometry Market, Emerging Technologies Empower Scientists, P8DD-30 (2016).
- A. Römpp, J.-P. Both, A. Brunelle, R.M.A. Heeren, O. Laprévote, B. Prideaux, A. Seyer, B. Spengler, M. Stoeckli, and D.F. Smith, Anal. Bioanal. Chem.407, 2329–2335 (2015).
- R.M.A. Heeren, Int. J. Mass Spectrom.377, 672–680 (2015).
- M. Vogeser and C. Seger, Clinical Biochemistry49, 947–954 (2016).
- A. Zask and G.A. Ellestad, Chirality27, 589–597 (2015).
- K.-H. Engel and G. Takeoka, Eds. Importance of Chirality to Flavor Compounds, ACS SYMPOSIUM SERIES 1212, Washington, DC (2015).
- J. Ye, M. Zhao, L. Niu, and W. Liu, Chem. Res. Toxicol.28, 325–338 (2015).
- W.A. Tao, F.C. Gozzo, and R.G. Cooks, Anal. Chem.73, 1692–1698 (2001).
- L. Wu, and F.G. Vogt, J. Pharm. Biomed. Anal.69, 133–147 (2012).
- D. Speybrouck and E. Lipka, J. Chrom. A,1467, 33–55 (2016).
- N.Berova, P. Polavarapu, K. Nakanishi, and R. Woody, Comprehensive Chiroptical Spectroscopy (John Wiley & Sons, 2012).
- S.R. Domingos, F. Hartl, W.J. Buma, and S. Woutersen, ChemPhysChem16, 3363–3373 (2015).
- U. Boesl and A. Kartouzian, Ann. Rev.Anal. Chem.9, 343–364 (2016).
- P. Horsch, G. Urbasch, and K.-M. Weitzel, Chirality24, 684–690 (2012).
- D. Patterson, M. Schnell, and J. Doyle, Nature497, 475–477 (2013).
- V.A. Shubert, D. Schmitz, C. Medcraft, A. Krin, D. Patterson, J.M. Doyle, and M. Schnell, J. Chem. Phys.142, 214201 (2015).
- B. Ritchie, Phys. Rev. A 13, 1411–1415 (1976).
- I. Powis, J. Phys. Chem. A104, 878–882 (2000).
- N. Böwering, T. Lischke, B. Schmidtke, N. Müller, T. Khalil, and U. Heinzmann, Phys. Rev. Lett.86, 1187–1190 (2001).
- G.A. Garcia, L. Nahon, M. Lebech, J.-C. Houver, D. Dowek, and I. Powis, J. Chem. Phys.119, 8781–8784 (2003).
- M. Dantus, M.H.M. Janssen, and A.H. Zewail, Chem. Phys. Lett.181, 281–287 (1991).
- J.A. Davies, J.E. LeClaire, R.E. Continetti, and C.C. Hayden, J. Chem. Phys. 111, 1–4 (1999).
- A.M. Rijs, M.H.M. Janssen, and C.C. Hayden, Phys. Rev. Lett.92, 123002 (2004).
- J. Ullrich, R. Moshammer, A. Dorn, R. Doerner, L.Ph.H. Schmidt, and H. Schmidt-Boecking, Rep. Prog. Phys. 66, 1463–1545 (2003).
- A. Vredenborg, W.G. Roeterdink, and M.H.M. Janssen, Rev. Sci. Instrum.79, 063108 (2008).
- A.I. Chichinin, K.-H. Gericke, S. Kauczok, and C. Maul, Int. Rev. Phys. Chem. 28, 607–680 (2009).
- A. Vredenborg, C.S. Lehmann, D. Irimia, W.G. Roeterdink, and M.H.M. Janssen, ChemPhysChem 12, 1459–1473 (2011).
- M.H.M. Janssen, The Reaction Microscope: Imaging and Pulse Shaping Control in Photodynamics, Lecture at CFEL/DESY, Hamburg, October 6, 2011.
- N. Bhargava Ram, C.S. Lehmann, and M.H.M. Janssen, XVIIIth International Conference on Ultrafast Phenomena,Lausanne (2012); EPJ Web of Conferences 41, 02029 (2013).
- A.T.J.B. Eppink and D.H. Parker, Rev. Sci. Instrum.68, 3477 (1997).
- U. Boesl, Mass Spec. Rev. 36, 86–109 (2017).
- K.L. Reid, Ann. Rev. Phys. Chem.54, 397–424 (2003).
- C.S. Lehmann, N.B. Ram, I. Powis, and M.H.M. Janssen, J. Chem. Phys.139, 234307 (2013).
- C. Lux, M. Wollenhaupt, T. Bolze, Q. Liang, J. Köhler, C. Sarpe, and T. Baumert, Angew. Chem. Int. Ed.51, 5001–5005 (2012).
- L. Nahon, G.A. Garcia, C.J. Harding, E. Mikajlo, and I. Powis, J. Chem. Phys.125, 114309 (2006).
- L. Nahon, L. Nag, G.A. Garcia, L. Myrgorodska, U. Meierhenrich, S. Beaulieu, V. Wanie, V. Blanchet, R. Geneaux, and I. Powis, Phys. Chem. Chem. Phys. 18, 12696 (2016).
- M.M.R. Fanood, I. Powis, and M.H.M. Janssen, J. Phys. Chem. A118, 11541–11546 (2014).
- M.M.R. Fanood, M.H.M. Janssen, and I. Powis, Phys. Chem. Chem. Phys. 17, 8614–8617 (2015).
- M.M.R. Fanood, M.H.M. Janssen, and I. Powis, J. Chem. Phys.145, 124320 (2016).
- C. Lux, M. Wollenhaupt, C. Sarpe, and T. Baumert, ChemPhysChem 16, 115–137 (2015).
- C. Lux, A. Senftleben, C. Sarpe, M. Wollenhaupt, and T. Baumert, J. Phys. B: At. Mol. Opt. Phys. 49, 02LT01 (2016).
- L. Nahon, G.A. Garcia, and I. Powis, J. Elec. Spec. Rel. Phenom. 204, 322–334 (2015).
- M.M.R. Fanood, N.B. Ram, C.S. Lehmann, I. Powis, and M.H.M. Janssen, Nat. Comm. 6, 7511 (2015).
- M.M.R. Fanood, “Molecular Chirality Under the Reaction Microscope,” PhD thesis, VU University Amsterdam, dare.ubvu.vu.nl/handle/1871/53390 (2015).
- M.H.M. Janssen and I. Powis, Phys. Chem. Chem. Phys. 16, 856–871 (2014).
- M. Stener, D. Di Tommaso, G. Fronzoni, P. Decleva, and I. Powis, J. Chem. Phys.124, 024326 (2006).
- R.E. Goetz, T.A. Isaev, B. Nikoobakht, R. Berger, and C.P. Koch, J. Chem. Phys.146, 024306 (2017).
- I. Dreissigacker and M. Lein, Phys. Rev. A 89, 053406 (2014).
- A.N. Artemyev, A.D. Müller, D. Hochstuhl, and Ph.V. Demekhin, J. Chem. Phys. 142, 244105 (2015).
- A. Ferré, C. Handschin, M. Dumergue, F. Burgy, A. Comby, D. Descamps, B. Fabre, G.A. Garcia, R. Géneaux, L. Merceron, E. Mével, L. Nahon, S. Petit, B. Pons, D. Staedter, S. Weber, T. Ruchon, V. Blanchet, and Y. Mairesse, Nature Photonics 9, 93–98 (2015).
- O.Kfir, P. Grychtol, E. Turgut, R. Knut, D. Zusin, D. Popmintchev, T. Popmintchev, H. Nembach, J.M. Shaw, A. Fleischer, H. Kapteyn, M. Murnane, and O. Cohen, Nature Photonics9, 99–105 (2015).
- S. Beaulieu, A. Ferré, R. Géneaux, R. Canonge, D. Descamps, B. Fabre, N. Fedorov, F. Légaré, S. Petit, T. Ruchon, V. Blanchet, Y. Mairesse, and B. Pons, New J. Phys. 18, 102002 (2016).
- A.Comby, S. Beaulieu, M.Boggio-Pasqua, D. Descamps, F. Légaré, L. Nahon, S. Petit, B. Pons, B. Fabre, Y. Mairesse, and V. Blanchet, J. Phys. Chem. Lett.7, 4514–4519 (2016).
- S. Beaulieu, A. Comby, B. Fabre, D. Descamps, A. Ferre, G. Garcia, R. Geneaux, F. Legare, L. Nahon, S. Petit, T. Ruchon, B. Pons, V. Blanchet, and Y. Mairesse, Faraday Discuss. 194, 325–348 (2016).
- A. Kastner, C. Lux, T. Ring, S. Zìllighoven, C. Sarpe, A. Senftleben, and T. Baumert, ChemPhysChem17, 1119–1122 (2016).
- M.E. Fermann and I. Hartl, Nature Photonics 7, 868 (2013).
Maurice H.M. Janssen is the founder and CEO of MassSpecpecD BV, the Netherlands (www.massspecpecd.com) and former professor of Molecular Photodynamics at the Institute for Lasers, Life and Biophotonics Amsterdam, VU University Amsterdam, the Netherlands. Direct correspondence to: info@massspecpecd.com. Ivan Powis is professor of Physical Chemistry (www.chem.nott.ac.uk/IP_Home.phtml) at the School of Chemistry, University of Nottingham, UK and special advisor at MassSpecpecD BV. Direct correspondence to: I.Powis@nottingham.ac.uk

















New Study Reviews Chromatography Methods for Flavonoid Analysis
April 21st 2025Flavonoids are widely used metabolites that carry out various functions in different industries, such as food and cosmetics. Detecting, separating, and quantifying them in fruit species can be a complicated process.



University of Rouen-Normandy Scientists Explore Eco-Friendly Sampling Approach for GC-HRMS
April 17th 2025Root exudates—substances secreted by living plant roots—are challenging to sample, as they are typically extracted using artificial devices and can vary widely in both quantity and composition across plant species.

