Analytical Characterization of Biopharmaceuticals


Davy Guillarme

Protein biopharmaceuticals are emerging as an important class of drugs for the treatment of various diseases including cancer, inflammatory diseases, and autoimmune disorders. There is currently a wide variety of protein biopharmaceutical formats, but clearly the two most promising ones are monoclonal antibodies (mAbs) and antibody−drug conjugates (ADC). The success of these molecules is related to their obvious benefits in terms of safety and efficacy-more than 2000 biopharmaceuticals products are currently in clinical development, while 250 products have ever been approved for human use (1). So, the future of the biopharmaceutical market looks undoubtedly bright.
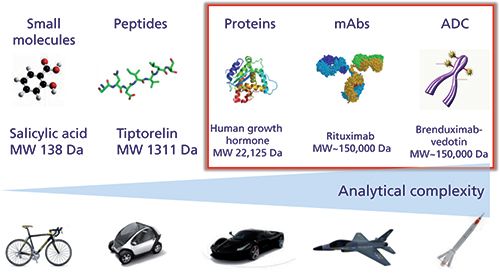
As illustrated in Figure 1, proteins, mAbs, and ADCs produced from living organisms have a complexity far exceeding that of small molecules and peptides produced from chemical synthesis. This additional complexity is related to some important sources of microheterogeneity observed in biopharmaceuticals that can occur during expression, purification, and long-term storage (that is, glycosylation, deamidation, oxidation, disulfide bridges scrambling, aggregation, and so on), as well as the fact that these small modifications must be characterized on a very large molecule. This inherent complexity can be well illustrated with mAbs, which possess a size of approximately 150 kDa, ± 1300 amino acids and several glycosylation sites. Despite the fact that only one single mAb is theoretically produced, there are always hundreds of possible variants that may exist, and all of them could contribute to the product efficacy and safety. Besides mAbs, ADCs further add to the complexity, since the heterogeneity of the original mAb is superimposed to the variability of the cytotoxic drug conjugation strategy. The ADC products are therefore often heterogeneous with respect to drug loading and its distribution on the mAb (2).

Figure 1: Analytical complexity for various classes of molecules.
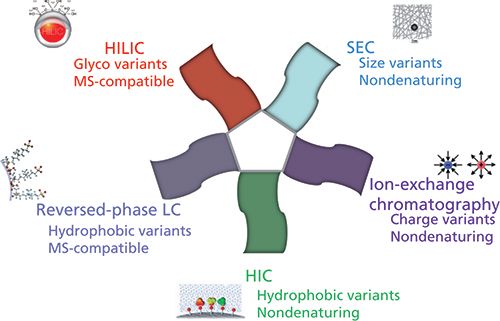
Today, there is a wide range of chromatographic approaches available for the successful characterization of mAbs and ADCs, as illustrated in Figure 2. Several nondenaturing approaches, including ion-exchange chromatography, size-exclusion chromatography (SEC), and hydrophobic-interaction chromatography (HIC), can be used for the separation of charge, size, and hydrophobic variants, respectively. However, these historical approaches, widely used by biochemists at the analytical and preparative scales, suffer from an inherent incompatibility with mass spectrometry (MS) because of the presence of a high salt concentration in the mobile phase. Therefore, some alternative MS-friendly strategies, such as reversed-phase liquid chromatography (LC) and hydrophilic-interaction chromatography (HILIC), have become more popular for the analytical characterization of proteins biopharmaceuticals.

Figure 2: Chromatographic approaches used for the analytical characterization of protein biopharmaceuticals.Figure 2: Chromatographic approaches used for the analytical characterization of protein biopharmaceuticals.
In the last few years, all the approaches described above have been drastically improved to meet the analytical requirements of very complex biopharmaceutical products. Some progress was made in terms of chromatographic instrumentation and columns. Among the most important advances, we can cite the following development and commercial introductions:
- bio-inert LC systems, to limit protein adsorption onto the injector, tubing, and ultraviolet (UV) cell,
- highly inert stationary phases, which are particularly useful in SEC and reversed-phase LC modes to limit adsorption, secondary interactions, and peak distortion,
- wide-pore HILIC columns for directly characterizing the glycosylation profile at the intact protein level, even for large proteins of up to >100 kDa,
- wide-pore columns packed with sub-2-µm fully porous particles or sub-4-µm core-shell particles to drastically improve the kinetic performance in reversed-phase LC, SEC, and HILIC modes,
- stationary phases compatible with very high temperatures (up to 90−100 °C), which is mandatory to limit protein adsorption in reversed-phase LC and improve peak shape, and
- easy-to-use buffers for pH gradients from 5.6 to 10.2, in replacement of the salt gradients commonly used in ion-exchange chromatography (3).
All these new features explain why the achievable performance of chromatographic approaches are better today than the electrophoretic approaches (in gel or capillary formats), which were the reference techniques for proteins analysis for a long time.
However, taking into account the inherent complexity of biopharmaceutical samples, all these efforts in terms of chromatographic improvements are still often insufficient to deal with such complex samples containing numerous closely related variants. This complexity is why there are still a lot of attempts to combine chromatographic strategies in a two-dimensional (2D) setup (comprehensive, selective 2D, or heart-cutting) both in academic laboratories and the biopharmaceutical industry. There are primarily two main reasons for using 2D-LC: First, complementary information on protein variants can be gained from the two dimensions of separation in a single analysis and high resolving power is expected; and second, enabling the hyphenation of nondenaturing approaches (ion-exchange chromatography, SEC, and HIC) to an MS detector to see the identity of the observed chromatographic peaks (4). Virtually, all of the above mentioned chromatographic techniques (that is, ion-exchange chromatography, SEC, HIC, reversed-phase LC, and HILIC) can be combined together. In practice, the second dimension of the 2D setup often includes reversed-phase LC (directly MS-compatible, thanks to the use of a volatile mobile phase) and sometimes SEC (possible separation of salts and proteins from the first dimension under nondenaturing conditions). Numerous 2D combinations have been tested for the characterization of mAbs and ADCs, in combination with MS detection:
- SEC × reversed-phase LC was used to characterize free drug species in ADC samples (5)
- ion exchange × reversed-phase LC was used to compare originator and biosimilar mAbs at the protein level (6), but also for characterizing ADC samples (7)
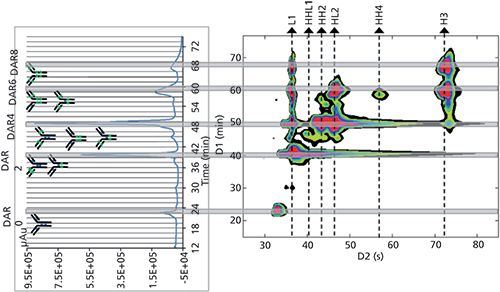
- HIC × reversed-phase LC was used to assess the drug loading, distribution, and conjugation sites of an ADC sample, as shown in Figure 3 (8)
- HILIC × reversed-phase LC, reversed-phase LC × reversed-phase LC, and ion exchange × reversed-phase LC modes were used to characterize mAbs at the peptide mapping level (9)

Figure 3: HIC × reversed-phase LC separation of an ADC sample (brentuximab vedotin). HIC separation is on the left side and a 2D-contour plot (HIC × reversed-phase LC) is on the right side. Adapted from reference 10, with permission from Elsevier.
Besides these examples, there are many other ways to combine chromatographic dimensions in a 2D-LC setup for characterizing mAb and ADC samples. With the increasing complexity of some new mAb formats (that is, bispecific mAbs [bsAbs], antibody-dual-drug conjugates [ADDCs], and so on), and the need to perform comparability studies between originator and biosimilar products, the future of 2D-LC looks brilliant.
Acknowledgments
The author wishes to thank the Swiss National Science Foundation for support (Grant 31003A_159494).
References
- G. Walsh, Nat. Biotechnol. 32(10), 992–1000 (2014).
- A. Beck, L. Goetsch, C. Dumontet, and N. Corvaïa, Nat. Rev. Drug Discov. 16(5), 315–337 (2017).
- S. Fekete, D. Guillarme, P. Sandra, and K. Sandra, Anal. Chem. 88, 480–507 (2016).
- D.R. Stoll, J. Danforth, K. Zhang, and A. Beck, J. Chromatogr. B 1032, 51–60 (2016).
- R.E. Bisdall, S.M. McCarthy, M.C. Janin-Bussat, M. Perez, J.F. Heuw, W. Chen, and A. Beck, mAbs 8(2), 306–317 (2016).
- M. Sorensen, D.C. Harmes, D.R. Stoll, G.O. Staples, S. Fekete, D. Guillarme, and A. Beck, mAbs 8(7), 1224–1234 (2016).
- K. Sandra, G. Vanhoenacker, I. Vandenheede, M. Steenbeke, M. Joseph, and P. Sandra, J. Chromatogr. B 1032, 119–130 (2016).
- M. Sarrut, A. Corgier, S. Fekete, D. Guillarme, D. Lascoux, M.C. Janin-Bussat, A. Beck, and S. Heinisch, J. Chromatogr. B 1032, 103–111 (2016).
- G. Vanhoenacker, I. Vandenheede, F. David, P. Sandra, and K. Sandra, Anal. Bioanal. Chem. 407, 355–366 (2015).
- M. Sarrut, S. Fekete, M.C. Janin-Bussat, O. Colas, D. Guillarme, A. Beck, and S. Heinisch, J. Chromatogr. B 1032, 91–102 (2016).
Davy Guillarme is with the School of Pharmaceutical Sciences at the University of Geneva, University of Lausanne in Geneva, Switzerland.








Regulatory Deadlines and Supply Chain Challenges Take Center Stage in Nitrosamine Discussion
April 10th 2025During an LCGC International peer exchange, Aloka Srinivasan, Mayank Bhanti, and Amber Burch discussed the regulatory deadlines and supply chain challenges that come with nitrosamine analysis.


Polysorbate Quantification and Degradation Analysis via LC and Charged Aerosol Detection
April 9th 2025Scientists from ThermoFisher Scientific published a review article in the Journal of Chromatography A that provided an overview of HPLC analysis using charged aerosol detection can help with polysorbate quantification.

