The Role of Chromatography in the Characterization and Analysis of Protein Therapeutic Drugs
Chromatography has taken a prominent place in the characterization and analysis of protein therapeutic drugs and today it plays a critical role in the biotechnology laboratory.
Chromatography has taken a prominent place in the characterization and analysis of protein therapeutic drugs and today it plays a critical role in the biotechnology laboratory. Although reversed-phase chromatography is the foremost chromatography technique used for this purpose, other techniques such as ion-exchange, size-exclusion, normal-phase, hydrophilic-interaction, and hydrophobic-interaction chromatography play very specific roles in characterizing and analyzing protein drugs.
Chromatography has long been an essential tool for protein purification. Dextran-based ion-exchange and size-exclusion columns have been instrumental in protein purification and characterization for decades. With the development of recombinant DNA proteins for therapeutic purposes there has been a need for careful, detailed characterization of these proteins, the changes and modifications that might occur, as well as sensitive determination of changes that actually do occur. Chromatography has taken its place as a premier technology in both the characterization and the analysis of recombinant protein therapeutic drugs (1).
Protein Modifications and Their Effect on Protein Therapeutic Drugs
The biological activity and, therefore, the therapeutic efficacy of protein drugs depends on the exact composition and three-dimensional structure of the protein. Any change to a protein (modification) may affect its efficacy as a therapeutic agent, although efficacy may not be affected by some protein modifications. The effect of a given modification depends on the protein and the nature and location of the modification. During development of a protein therapeutic drug it is necessary to fully characterize the protein, define what possible modifications may occur, define those modifications that do affect efficacy (critical quality attributes), and develop methods to analyze such modifications. Changes occur to proteins from the beginning of their assembly. Some changes may be needed for the protein to be properly formed into an active tertiary structure such as glycosylation and disulphide bonds, which are critical in maintaining the proper structure. Glycosylation attaches sugar chains to either asparagine or serine residues. These sugar chains affect protein folding and assist in maintaining correct tertiary protein structure. Glycosylation structure is complicated and is affected by the cell lines used for protein expression as well as other aspects of the expression system. It is vital to characterize and monitor glycosylation patterns. Disulphide bonds formed between cysteine residues are essential for formation and maintenance of proper tertiary structure. These must be characterized as to their locations in the primary structure of a protein and also must be monitored to ensure that a protein remains active.
Other modifications may affect protein binding and reduce activity such as chemical deamidation — the conversion of an asparagine residue into an aspartic or isoaspartic acid residue under conditions of high temperature or high pH — and oxidation of methionine residues under chemical oxidative conditions to methione sulphoxide. These two modifications may affect activity if found near catalytic or binding sites, but may have no effect if found distant from sites of activity.
Some modifications are engineered into proteins to form more effective drugs. The addition of polyethylene glycol to proteins — pegylation — usually extends the duration of protein activity in the blood stream several times by reducing the rate at which the protein is removed from the blood stream. Monoclonal antibodies (mAbs) that bind to specific receptors on the surface of tumour cells can be modified by the addition of highly toxic drugs to the monoclonal antibody, creating a highly specific, effective reagent that targets and kills specific tumour cells. Engineered modifications must be analyzed and monitored.
Proteins may lose tertiary structure by variations in pH, temperature, or the presence of certain reagents. Such "denatured" proteins lose biological activity and pharmacological potency. Proteins may also form "aggregates," where multiple proteins self-associate into larger complexes that usually reduce activity and may engender immune responses in patients.
Reversed-Phase HPLC
The most powerful and useful chromatography technique used for analysis and characterization of proteins drugs is reversed-phase high performance liquid chromatography (HPLC). This technique is most commonly used to separate, identify, and measure peptides that result from the enzymatic digestion of the therapeutic protein (peptide map). Although a single modification on a large, many kilodalton protein may cause enough of a change in the retention of the protein in reversed-phase HPLC to differentiate the modified protein from the native protein, the change is often too small to cause a measurable change in retention. By digesting a protein into smaller pieces — typically from a very few amino acids to 20–40 amino acids — the modification now makes a much larger change in the retention of the modified peptide relative to the native peptide than for the intact protein, thus allowing easier detection and quantitation of the modification. Figure 1 shows the effect on peptide retention of the addition of a methionine residue to human growth hormone, which occurs when the protein is expressed in E. coli bacteria. Such peptide maps are the basis of many chromatography methods for both characterization and analysis of therapeutic proteins. Among protein modifications that are easily detected and measured by peptide maps are deamidation of an asparagine residue, oxidation of a methionine residue, and conversion of an N-terminal glutamine residue to cyclized glutamate. These modifications may diminish the efficacy of a therapeutic protein or otherwise affect its potency as a therapeutic agent. Other protein characteristics that can be monitored by peptide maps include disulphide bonds between two cysteine residues and exact locations of glycosylation. Reversed-phase HPLC is widely used in the analysis and characterization of therapeutic proteins because the list of modifications that can be characterized, identified, or quantified by this technique is very long.


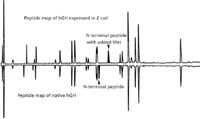
Figure 1: Effect of the addition of a methionine residue to human growth hormone because of expression in E. coli on the relative retention of a peptide in a peptide map. Met is somewhat hydrophobic so the peptide containing Met is retained longer than the (native) peptide absent the Met residue. Adapted from reference 1.
The Separation of Polypeptides by Reversed-Phase HPLC
Although reversed-phase HPLC has been known and practiced for several decades, it was initially considered impractical for separating large molecules such as proteins or even peptides. Early attempts at protein or peptide separations by reversed-phase HPLC resulted in poor peak shapes and separations. The introduction of larger-pore silica (average pore size: 150–300 Å) for chromatography columns in the early 1980s provided the first really high resolution of large polypeptides (2,3). This discovery coincided with the early days of recombinant DNA protein development and the two grew hand in hand. The use of large pore silica opened the door to rigorous development of optimal conditions for polypeptide separations and, today, such optimized conditions coupled with high quality reversed-phase HPLC separation columns result in separations that are highly effective in characterizing and monitoring therapeutic drugs.

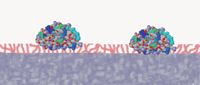
Figure 2: Polypeptides are large molecules that adsorb strongly to the hydrophobic surface and desorb only at a very specific solvent strength. A portion of the polypeptide adsorbs to the surface (stationary phase), and some of the molecule remains in contact with the mobile phase. See reference 3.
Larger pores allow the entrance of the larger protein and peptide molecules into the interior of the pores where the bulk of the adsorptive surface causing separation is found. The relatively large polypeptide then adsorbs to the surface of the modified, hydrophobic surface (Figure 2). Large polypeptides are more strongly bound to the hydrophobic surface than smaller molecules and do not desorb from the surface until a stronger solvent (for example, more hydrophobic solvent) enters the column and pores. Once desorbed, the polypeptide appears to interact minimally with the surface of the silica during its transport down the column. The amount of separation is essentially determined by the strength of that initial adsorption, which in turn is determined by the size and hydrophobicity of the portion of the polypeptide that actually interacts with the hydrophobic surface. This portion of the molecule is called the hydrophobic foot (4) and is the key to polypeptide separations. Because of the large size of the polypeptide molecule, only a portion will interact with and adsorb to the hydrophobic surface. A portion of the polypeptide will be in contact with the mobile phase. Even very small changes to the hydrophobic foot cause a change in the retention of molecules in reversed-phase HPLC resulting in separation. This accounts for the ability of reversed-phase HPLC to separate proteins or peptides that differ in even very small ways from a native or reference protein or peptide. In the separation of two insulin variants shown in Figure 3, a difference of only a single methyl group (rabbit insulin has a threonine where human insulin has a serine, a difference of a single methyl group) which represents a difference of 15 MW in a protein of about 5700 MW, and yet reversed-phase HPLC is able to separate these two proteins (5). The ability of reversed-phase HPLC to separate such very similar polypeptides is at the heart of its use in the characterization and analysis of protein therapeutic drugs.

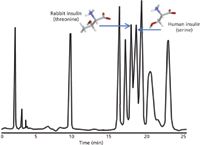
Figure 3: The ability of reversed-phase HPLC to separate very similar polypeptides is illustrated by the separation of two insulin variants (human and rabbit) that differ by a single amino acid, which in turn differ by a single methyl group. Adapted from reference 2.
Conditions for Polypeptide Separations by Reversed-Phase HPLC
The separation column is based on small, porous silica particles of 1.7–5 μm diameter. Most protein therapeutic drug separations use 5-μm silica particles, however, smaller particle columns are increasing in usage because of shorter analysis times and, sometimes, higher resolution (6) for separation of mAbs using superficially porous silica particles. The best pore size for polypeptides is normally between 150 Å and 300 Å, although very small peptides, and sometimes enzymatic digests, can be satisfactorily separated using columns of 100-Å pore size silica. The polar surface of the silica particles is modified by short (4–18 carbon atoms) aliphatic organic chains. Longer chain groups (for example, 18 carbons) are usually best to separate small peptides and protease digests of proteins while smaller hydrophobic groups (such as four carbons) are best for retention and separation of larger peptides (>15–20 residues), proteins, or hydrophobic polypeptides.


Table 1: Chromatography techniques commonly used for protein modification analysis.
The mobile phase consists of water, an organic solvent (nearly always acetonitrile), and an ion-pair reagent. Although acetonitrile is the most commonly used organic solvent when separating polypeptides, adding a small (1–5%) percentage of isopropanol can be an aid in the separation of hydrophobic polypeptides or when sample recovery is low.
The ion-pair reagent used is almost always trifluoroacetic acid, which evaporates well, is relatively UV transparent, and results in sharp polypeptide peaks and good resolution. Trifluoroacetic acid is usually added to the mobile phase at a concentration of about 0.1%. With peptide maps, the concentration of the trifluoroacetic acid may affect resolution between peptide pairs to a small extent and trifluoroacetic acid concentrations of 0.1–0.5% have been used to achieve slightly better resolution of critical pairs of peptides. When using electrospray mass spectrometry (MS) with reversed-phase HPLC, the use of trifluoroacetic acid results in a much lower signal than obtained in its absence. The signal loss depends on the concentration of trifluoroacetic acid, and low concentrations of 0.01–0.05% result in relatively little signal loss; however, resolution and peak shape may suffer at the lower concentrations of trifluoroacetic acid. It has been shown, however, that very low concentrations of trifluoroacetic acid result in good peak shape and resolution if very high purity silica (Type B) columns are used. Consequently, many manufacturers offer very high purity columns specifically for use with trifluoroacetic acid and reversed-phase HPLC–MS. Formic acid has also been used by some polypeptide chromatographers to avoid the signal loss in MS when using trifluoroacetic acid, although some chromatographers have found decreased resolution with formic acid, so it is usually reserved for MS applications. Other ion-pair reagents such as heptafluorobutyric or pentafluoroproprionic acid have been used on occasion, particularly for more-hydrophobic polypeptides.
Gradient elution is always used in polypeptide separations. The organic solvent concentration is low (0–5%) at the beginning of the separation and is increased slowly over the course of the separation. Because of the adsorption mechanism of polypeptides on the hydrophobic surface, very small changes in organic solvent concentration affect the retention of a polypeptide, so very small changes in the organic solvent concentration (gradient slope) can have a significant effect on retention and resolution. Shallow gradient slopes of 0.1–0.5% are commonly used, resulting in separation times of 30 min to as high as 180 min for separating complex mixtures of peptides, such as enzymatic digests of very large proteins. Occasionally, the gradient slope will affect the resolution of peptide pairs in unexpected ways. Although reductions in gradient slope (resulting in increased elution times) generally lead to increased resolution, because of differences in the thermodynamics of adsorption of some peptides, the resolution of some peptide pairs may decrease (7) with decreasing gradient slope. Gradient slope, then, must be carefully optimized to achieve optimal resolution of the peptides in a protease digest in the best time.
The column temperature also affects the resolution of some peptide pairs because of differences in the thermodynamics of adsorption of some peptides (Figure 4) (8). Because temperature can affect the relative retention of peptides, several temperatures should be tried when developing a peptide map separation with careful monitoring of the effect of temperature changes. The optimized column temperature is a very important element in establishing an analysis method and its validation. High temperatures often improve peak shape and resolution when analyzing large polypeptides such as mAbs (9).

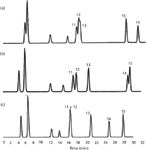
Figure 4: Slight differences in the way that peptides adsorb onto the reversed-phase HPLC surface results in variations in relative retention of some peptides at (a) 20 °C, (b) 40 °C, and (c) 60 °C. This leads to changes in resolution between some peptide pairs in a reversed-phase HPLC peptide map. In this example of the peptide map of human growth hormone, peptides 11, 12, and 13 are poorly separated at 20 °C, are much better separated at 40 °C, but are coeluted at 60 °C. Peptide 15 is eluted before peptide 14 at 20 °C, slightly later at 40 °C, and significantly later at 60 °C. Temperature often has a significant effect on peptide resolution and must be optimized when developing a peptide map separation. Adapted from reference 7.
Detection of polypeptides in reversed-phase HPLC is normally done at or near 214 nm. This wavelength offers optimal detection of polypeptides. During a typical gradient to separate peptides, the baseline may rise because of a shift in adsorption at this wavelength by trifluoroacetic acid as the dielectric constant of the solvent changes. This can be compensated by reducing the concentration of trifluoroacetic acid in the organic solvent by 10–15% relative to the aqueous solvent.
Ion-Exchange Chromatography
Ion-exchange chromatography has long been a useful tool in the purification of proteins and remains a mainstay in the purification of therapeutic protein drugs. However, ion-exchange chromatography does not have the resolving power of reversed-phase chromatography, and is usually not able to separate small differences between protein molecules. Despite this, because ion-exchange chromatography separates by a different mechanism than reversed-phase HPLC — it separates proteins by charge rather than hydrophobicity — it can perform certain separations better than reversed-phase HPLC. Proteins bind strongly to the ionized surface of ion-exchange materials by the opposite charges found on the protein. Cation-exchange chromatography, with particles containing negative charges (sulphonic or carboxylic acid groups), binds a protein by the positive charges of lysine and asparagine (also the amino terminus). Anion-exchange chromatography uses particles with positive charges and binds the negatively charged amino acids aspartic and glutamic acids (as well as the carboxy terminus of a protein). Although ion-exchange chromatography lacks the high resolving power of reversed-phase HPLC, it has found use in monitoring modifications to proteins that change the overall charge on a protein (called charge variants). This includes modifications such as deamidation, which adds a negative charge to a protein, converting the neutral asparagine to a negatively charged aspartic (or isoaspartic) acid group. Other charge variants are formed when lysine is enzymatically removed from the carboxy terminus of an antibody heavy chain; when N-terminal glutamine cyclizes with the amino terminus, thus losing a positive charge; when molecules such as polyethylene glycol are added to primary amines in a protein, thus losing a positive charge; or when acidic sugars (such as sialic acid) are added to the ends of glycan chains. These charge variants can be effectively monitored using ion-exchange chromatography (10). Conditions are simple; the ion exchange materials have charged (negative or positive) groups on their surface to which the proteins bind. Elution is accomplished with a simple linear increase in salt — typically sodium chloride — concentrations in the mobile phase. Gradients from 0–2 M salt suffice to elute almost all proteins. Separation and resolution of proteins is very sensitive to the mobile-phase pH and the optimum pH must be empirically determined for each protein.
The use of ion-exchange and reversed-phase HPLC in multidimensional LC × LC techniques combines the strengths of the two different mechanisms and results in very high resolution separations. For example, the use of a cation-exchange column as the first stage will separate peptides on the basis of charge and individual peptides with similar charge can be further separated by using a reversed-phase second column. The operation can be run with off-line collection of the effluents from column 1 with manual reinjection onto column 2. Some workers prefer on-line coupling of the two modes by the use of high-pressure switching valves after column 1 to direct the effluent to column 2 and allow automation of the entire operation.
Size-Exclusion Chromatography
Another form of chromatography long used in protein purification is size-exclusion chromatography (SEC), sometimes called gel filtration when using an aqueous mobile phase. SEC separates molecules, particularly proteins, by differences in three-dimensional size, including water bound to the protein (hydrodynamic volume). Although the resolution between proteins of different sizes is not very good, size exclusion has found an important place in protein therapeutic drug characterization and analysis by separating monomeric proteins from aggregates, a crucial factor in protein analysis. Size exclusion is also an useful technique in monitoring denaturation if that occurs. Columns with different fractionation ranges can be obtained and the mobile phase is designed to simply solubilize the protein very well. The use of SEC in the characterization of proteins and their aggregates is covered elsewhere in this issue (11).
Chromatographic Analysis of Glycosylation
The nature and structure of sugar chains attached to proteins (glycosylation) must be regularly monitored in protein therapeutic drug batches. This ensures reproducibility of the production process and minimizes any effects that changes in glycosylation may have on protein activity and therefore drug efficacy. For many years the technique of high-pH anion-exchange chromatography (HPAEC) has been used to monitor glycosylation. In recent years other chromatographic techniques and nonchromatography techniques (such as capillary electrophoresis and MS) have been developed to provide better or more sensitive analysis of glycosylation patterns. In HPAEC, the mobile-phase pH is adjusted to a pH of 11–13, thus ionizing the hydroxyl groups on the sugars. These are then separated by anion-exchange chromatography using special instrumentation designed to operate under these corrosive conditions. Because sugars do not contain UV chromaphores, detection is done by electrochemical means. To avoid the corrosive conditions of HPAEC, other chromatography techniques have been developed for glycan analysis. In most cases, these techniques require the use of sugar derivatization with fluorescent molecules, such as anthranilic acid, to facilitate detection. Derivatized glycans can be detected with high sensitivity by fluoresence and can be separated by a variety of chromatographic techniques. Reversed-phase chromatography of derivatized glycans is effective at separating and quantitating glycans (12). Normal-phase chromatography is also effective at separating glycans (13). Normal-phase chromatography relies on the hydrogen bonding of the polar sugar hydroxyl groups to amine groups on the surface of the chromatographic particles for separation. Reversed-phase HPLC and normal-phase techniques have the advantage of being able to be coupled with MS, offering additional information on the nature of the glycans. A more recent addition to the chromatographic lineup for glycan analysis is hydrophilic interaction chromatography (HILIC). This technique uses polar particles (often bare silica) with a mobile phase containing high concentrations of acetonitrile, with relatively low concentrations of water containing salt. The aqueous solution adsorbs onto the particle surface forming a layer of very hydrophilic liquid. Polar compounds such as glycans partition from the relatively organic mobile phase into the relative hydrophilic phase adsorbed to the particle surface. Elution is usually by means of an increase in the water content of the mobile phase and a subsequent decrease in the organic phase over time. Glycans, being very polar molecules, separate very well by this technique and effort has gone into developing a practical framework by which to identify and characterize the glycans that are separated by small-particle HILIC columns (Figure 5).

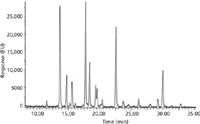
Figure 5: Glycans are very polar molecules and can be separated and analyzed by hydrophilic interaction chromatography. This chromatogram shows the separation of glycans present on the protein bovine fetuin. Data courtesy of Waters Associates.
Conclusion
Chromatography has taken a prominent place in the characterization and analysis of protein therapeutic drugs and today plays a critical role in the biotechnology laboratory. Although reversed-phase is the foremost chromatography technique used for this purpose, other techniques such as ion exchange, size exclusion, normal phase, HILIC, and HIC play very specific roles in characterizing and analyzing protein drugs.
David Carr is with Bioanalytical Technologies. He has been involved with HPLC for more than 40 years. He has worked with the biotechnology industry for many years in the characterization and analysis of protein therapeutics and is the author of the booklet "The Handbook of Analysis and Purification of Proteins and Peptides by Reversed-Phase HPLC". For the past 16 years he has taught the class "The Analysis and Characterization of Protein Therapeutic Drugs for Bioanalytical Technologies" (www.bioanalyticaltech.com) to scientists from most of the major biotechnology firms and many smaller biotech companies. Direct correspondence to: davidc.carr@verizon.net
References
(1) R.L. Garnick, N.J. Solli, and P.A. Papa, Anal. Chem. 60(23), 2546–2557 (1988).
(2) J.D. Pearson, N.T. Lin, and F. Regnier, Anal. Biochem. 124(1), 217–30 (1982).
(3) J. Rivier et al., J. Chromatogr. 268, 1121 (1983).
(4) X. Geng and F. Regnier, J. Chromatogr. 296, 15–30 (1984).
(5) J. Rivier and R. McClintock, J. Chrom. 268, 112–119 (1983).
(6) S. Fekete, S. Rudaz, J. Fekete, and D. Guillarme, J. Pharm. Biomed. Anal. 70, 158–168 (2012).
(7) R.C. Chloupek, W.S. Hancock, and L.R. Snyder, J. Chromatogr. 594, 65–73 (1992).
(8) W.S. Hancock, R.C. Chloupek, J.J. Kirkland, and L.R. Snyder, J. Chromatogr. 686, 31–43 (1994).
(9) S. Fekete, S. Rudaz, J-L. Veuyhey, and D. Guillarme, J. Sep. Sci. 35, 3113–3123 (2012).
(10) Y. Lyubarskaya, D. House, J. Woodard, and R. Mhatre, Anal. Biochem. 348(1), 24–39 (2005).
(11) L. Lloyd, LCGC Europe 27(s11), 25–29 (2014).
(12) X. Chen, Y.D. Liu, and G. Flynn, Glycobiology 19(3), 240–249 (2009).
(13) K.R. Anumula and S.T. Dhume, Glycobiology 8(7), 685–694 (1998).










New TRC Facility Accelerates Innovation and Delivery
April 25th 2025We’ve expanded our capabilities with a state-of-the-art, 200,000 sq ft TRC facility in Toronto, completed in 2024 and staffed by over 100 PhD- and MSc-level scientists. This investment enables the development of more innovative compounds, a broader catalogue and custom offering, and streamlined operations for faster delivery. • Our extensive range of over 100,000 high-quality research chemicals—including APIs, metabolites, and impurities in both native and stable isotope-labelled forms—provides essential tools for uncovering molecular disease mechanisms and exploring new opportunities for therapeutic intervention.
New Guide: Characterising Impurity Standards – What Defines “Good Enough?”
April 25th 2025Impurity reference standards (IRSs) are essential for accurately identifying and quantifying impurities in pharmaceutical development and manufacturing. Yet, with limited regulatory guidance on how much characterisation is truly required for different applications, selecting the right standard can be challenging. To help, LGC has developed a new interactive multimedia guide, packed with expert insights to support your decision-making and give you greater confidence when choosing the right IRS for your specific needs.

