Method for Heart-Cut Analysis Using Nanoacquity UPLC With 2D Technology for Proteomic Samples
A biomarker is measured as an indicator of normal biological processes, pathogenic processes, or pharmacologic responses to therapeutic intervention. Many times, a putative biomarker is a protein or peptide that is expressed at a relatively low level compared to the surrounding proteome. The constitutive or housekeeping proteins are present in concentrations that are orders of magnitude above the protein of interest, which makes identification and quantitation difficult. In order to validate a candidate biomarker, many samples need to be analyzed to prove that the same analytes are reproducibly identified and are changing in a statistically significant manner due to a perturbation.
Martha D. Stapels, Keith E. Fadgen, and James I. Langridge, Waters Corporation, Milford, MA, U.S.
INTRODUCTION
A biomarker is measured as an indicator of normal biological processes, pathogenic processes, or pharmacologic responses to therapeutic intervention. Many times, a putative biomarker is a protein or peptide that is expressed at a relatively low level compared to the surrounding proteome. The constitutive or housekeeping proteins are present in concentrations that are orders of magnitude above the protein of interest, which makes identification and quantitation difficult. In order to validate a candidate biomarker, many samples need to be analyzed to prove that the same analytes are reproducibly identified and are changing in a statistically significant manner due to a perturbation.
Two-dimensional (2D) chromatography is often used to separate peptides from proteomic samples in a biomarker discovery workflow. 2D chromatography of tryptic peptides has traditionally been performed using strong cation exchange (SCX) followed by reversed-phase (RP) chromatography, due to the orthogonal nature of the separation mechanisms of these two techniques. The major disadvantage of this approach is that peptides are often split across first dimension SCX fractions due to the relatively low resolution of peptides on SCX material.
A highly reproducible method for performing online 2D chromatography with mass spectrometry has been developed where peptides are separated by RP chromatography at high pH in the first dimension, followed by an orthogonal separation at low pH in the second dimension. An online dilution of the effluent was performed after the first dimension to ensure no peptides were lost during trapping prior to the second dimension. For targeted biomarker validation, running an entire 2D experiment would be time consuming given the limited number of target molecules that might need to be monitored and the number samples in a typical validation experiment. A preferred approach is to elute the targeted peptides in one fraction in a heart-cut manner. This application note will illustrate the application of online high/low pH RP/RP chromatography for heart-cut analysis.
EXPERIMENTAL
LC/MS conditions



Sample preparation and loading: Waters MassPREP™ E. coli digestion standard (2.4 μg) with either MassPREP Protein Digestion Standard Mix 1 or Mix 2 was analyzed. In each case, three replicate injections were performed.
Online dilution with RP/RP: To maximize sample recovery on the second-dimension trap column from the organic-containing fractions, an aqueous flow was delivered with the second dimension pump, and mixed with the eluted fraction prior to trapping, as shown in Figure 1B.

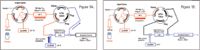
Figure 1. Fluidic layout for the 2D-nanoACQUITY UPLC System for high/low pH RP/RP analysis with online dilution. Two gradient pumps (nanoBSM) were utilized for (A) sample injection and fractionation, as well as (B) analytical gradient delivery.
Heart-cut method: Peptides from unwanted fractions were washed away by loading the sample at an ACN concentration just below the fraction of interest. During dilution, the second dimension pump delivered a high ACN concentration to prevent peptides sticking onto the trap column. To retain the desired fraction, aqueous conditions were utilized on the second dimension dilution pump.
Data acquisition and processing: LC-MSE data from the individual chromatograms were processed separately and subsequently merged into one file prior to database searching with ProteinLynx Global Server™ Software (PLGS 2.4 with IdentityE Informatics). An E. coli database with an equal number of random sequences concatenated onto it was used for the searches to limit the false positive rate to less than 4%.


Figure 2. Chromatograms for five fraction 2D-analysis of Mix 1 in E. coli.
RESULTS AND DISCUSSION
2D separations
The 2D separation of an E. coli tryptic digest containing Mix 1 is shown in Figure 2. Organic concentrations for each step were selected to ensure nearly equivalent peptide load and MS intensity for each second-dimension run. Due to the nature of RP gradients, the majority of peptides were only identified in one fraction (86.2% for Mix 1 in E. coli, and 87.3% for Mix 2 in E. coli). Figure 3 depicts the number of E. coli proteins identified in two of three replicate 2D experiments. The data point in red is 50 fmol ADH, to show the level of the spiked-in proteins relative to the background. In three replicate analyses, the average relative standard deviation of the protein fmol amount was 15%.

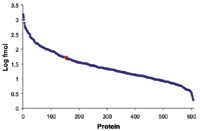
Figure 3. Absolute amount2 of proteins detected from a 2D 5-fraction analysis of Mix 1 spiked in E. coli.
Heart-cut separations
Figure 4 shows the replicate analyses of the heart-cut analysis of the E. coli sample containing Mix 1, where peptides were eluted with 20.8% ACN from the first-dimension column (the equivalent of fraction 4). The elution profile of the second-dimension is the same as in the full 2D-analysis, with peptides eluting from 25 to 85 minutes.


Figure 4. Replicate heart-cut analyses of fraction 4 of Mix 1 in E. coli.
Qualitative reproducibility
The 2D method was run in triplicate on both Mix 1 and 2 spiked into E. coli. It was found that 607 and 593 proteins (87% of the total number of identified proteins) replicated in two of the three analyses of each of the samples, respectively. The comparison of the identified peptides from the fourth fraction of the 2D-experiment with the heart-cut experiment is shown in Figure 5. In order to ensure that the peptides of interest are found in the heart-cut fractions, the acetonitrile steps can be widened slightly (0.5% on each side of the step) without significantly altering the separation efficiency.

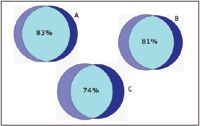
Figure 5. Venn diagrams showing (A) the overlap of highly confident peptides identified in fraction 4 from replicate 2D experiments, (B) the overlap of peptides identified from replicate heart-cuts, and (C) the overlap of peptides identified from the two different methods.
Quantitative reproducibility
Peak areas of peptides from digested protein standards were measured to test the ability of the system to perform label-free quantitation, especially with a heart-cut analysis of only one fraction. Figures 6 and 7 show the results from this analysis. The average measured protein ratios were 7.30, 0.52, 1.00, and 2.20 for the 2D analysis; and 6.70, 0.53, 1.00, and 1.90 for the heart-cut analysis for BSA, glycogen phosphorylase (Phos B), alcohol dehydrogenase (ADH), and enolase, respectively. The measured ratios were within 6.3% and 7.7% of the expected theoretical values on average for the 2D and heart-cut methods. The peak areas were very consistent between the two methods with more peptides and greater intensity for common peptides identified in the 2D-method, as expected.


Figure 6. Intensity of peptides to the four standards in Mixes 1 and 2 found in the 5 fraction 2D-LC method. The theoretical protein ratios are shown.
CONCLUSION
A method for heart-cut analysis has been implemented on the nanoACQUITY UPLC System and can yield the same qualitative and quantitative information that is obtained in entire 2D analyses, in a fraction of the time. The 2D experiment shown here (two samples with five fractions run in triplicate) took 60 hours of instrument time, while the heart-cut equivalent took 15 hours to complete. Since the majority of peptides in 2D RP/RP analyses were only found in one fraction and they were reproducibly found in the same fraction in replicate analyses, this technique is well-suited to subsequent heart-cut analyses for targeted proteomics and biomarker verification studies. The results from the analysis of standard proteins spiked into E. coli show that label-free relative quantitation works well with both methods.


Figure 7. Intensity of peptides to the four protein standards in Mixes 1 and 2 found in the fraction 4 from the first dimension using the heart-cut method. The theoretical protein ratios are shown.
References
1. M. Gilar, et.al., J. Sep. Sci. 2005, 28, 1694-1703.
2. J. Silva, et.al., Mol. Cell Proteomics, 2006, 144-156.








Common Challenges in Nitrosamine Analysis: An LCGC International Peer Exchange
April 15th 2025A recent roundtable discussion featuring Aloka Srinivasan of Raaha, Mayank Bhanti of the United States Pharmacopeia (USP), and Amber Burch of Purisys discussed the challenges surrounding nitrosamine analysis in pharmaceuticals.


Extracting Estrogenic Hormones Using Rotating Disk and Modified Clays
April 14th 2025University of Caldas and University of Chile researchers extracted estrogenic hormones from wastewater samples using rotating disk sorption extraction. After extraction, the concentrated analytes were measured using liquid chromatography coupled with photodiode array detection (HPLC-PDA).

