The LCGC Blog: Best Practice in HPLC Eluent Design
I think its fair to say that there has been a paradigm change in the way we approach mobile-phase design for HPLC in recent years.
I think it's fair to say that there has been a paradigm change in the way we approach mobile-phase design for HPLC in recent years. The trends are for more simple linear binary gradients with fewer additives (buffers, sacrificial bases, exotic pH adjusting reagents) linked in part to an improvement in column stationary phase design, the requirements for volatile buffers to allow API-MS detector compatibility, but also closely aligned to the requirements for the inherent robustness that accompanies more straightforward eluent systems.
While the new mobile phases, including 0.1% TFA, 0.1% formic acid and 10 mM ammonia, certainly deliver improvements, we are still missing several tricks to further improve the quality of the chromatographic output that can be achieved via the use of the simpler eluent systems.
I often see chromatograms with sloping, noisy baselines and sub-optimal sensitivity for separations that use these more straightforward eluents. What follows are some key tips in order to make the very best use of these new approaches to eluent design.
For those who are developing new HPLC methods, it’s important to realize that for certain separations, the ionic strength of the mobile phase can alter the selectivity obtained and this will not only depend upon the analyte chemistry but also on the nature of the bonded phase and silica substrate. In short, any column screening process that is undertaken, may be confounded by secondary ionic strength gradients (higher to lower ionic strength) that occur when the buffer is added to the aqueous component (Mobile Phase A) only.
Figure 1 shows the difference in selectivity obtained in an isocratic separation when the buffer strength is altered.


Figure 1: Changing separation selectivity with altered buffer strength in an isocratic separation of food additives
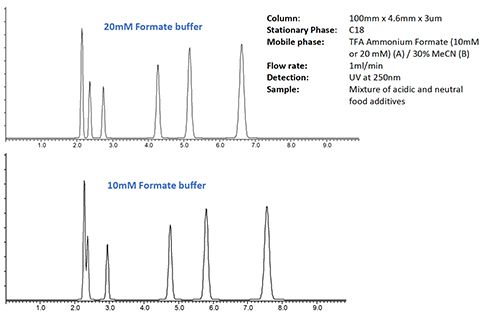
It should be noted that both separations in Figure 1 are carried out at exactly the same pH. The only difference between the two separations is the concentration of the ammonium acetate buffer.
It is also important to note that when altering eluent ionic strength, the magnitude of selectivity changes will depend upon the nature of the interactions between the analyte and the bonded-phase ligand or stationary-phase surface. Just to reiterate, during method development, when column selectivity is being evaluated, it’s important to ensure that the results are not confounded due to variability arising from susceptibility to changing ionic strength.
Figure 2 shows screening experiments being undertaken on two different stationary phases with ionic strength (nominally 10 mM ammonium acetate) varying over the gradient (5–60%), with only the aqueous component (mobile phase A) being ionic strength adjusted. This contrasted with the same experiment where the ionic strength is kept constant via the use of a ternary pumping system in which a constant 5% of a 200 mM ammonium acetate is delivered to the pump.

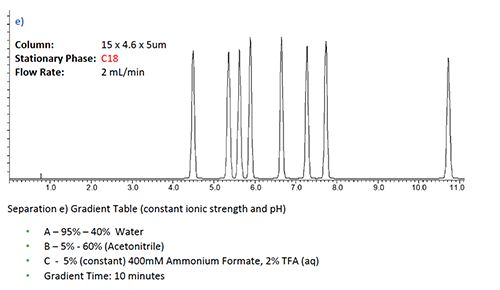
Figure 2: Selectivity affects in gradient HPLC analysis with various buffered eluent systems and column chemistries for a sample of basic and neutral pharmaceutical analytes. a) & b) variable ionic strength variable pH gradient with C18 and C8 stationary phase respectively, c) & d) same separation under constant ionic strength conditions, separation e) C18 separation under constant ionic strength and pH conditions.
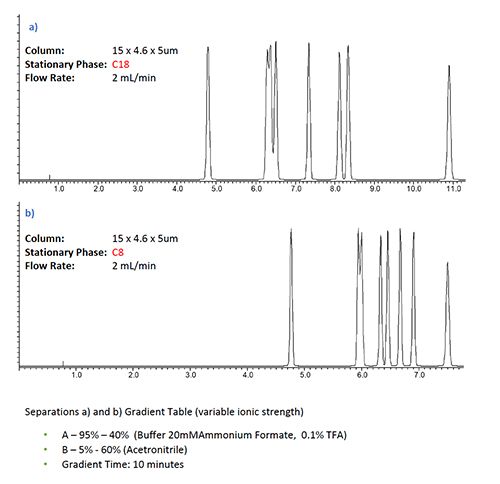

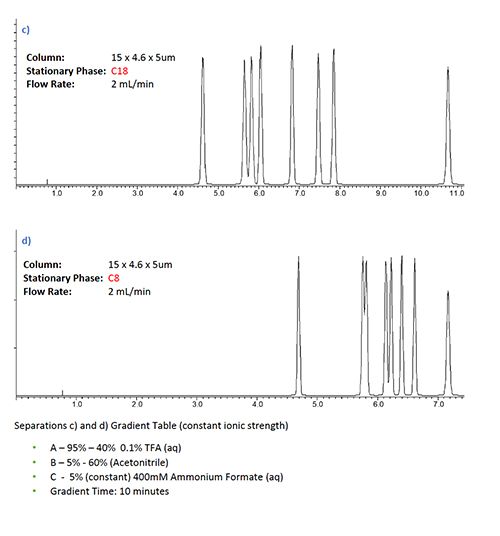

Figure 2: Selectivity affects in gradient HPLC analysis with various buffered eluent systems and column chemistries for a sample of basic and neutral pharmaceutical analytes. a) & b) variable ionic strength variable pH gradient with C18 and C8 stationary phase respectively, c) & d) same separation under constant ionic strength conditions, separation e) C18 separation under constant ionic strength and pH conditions.
From Figure 2 it is clear that there are some marked differences in the selectivity between the ionic strength gradient separations (a,b) and the constant ionic strength separations (c,d). It is also worthy of note that the constant ionic strength is easy to achieve using a simple ternary gradient. While some QC laboratories prefer to use binary pumping equipment due to perceived improvements in reproducibility and simplicity of transfer between different instrument types, in my experience, with only a little care, the use of a quaternary pumping system can be just as robust, repeatable, and transferable as a gradient system.
It is also worthy of note that the differences between the selectivity in variable and constant ionic strength methods are more obvious for the C18 than the C8 phase.
Figure 2e) indicates that there is also a change in selectivity when the pH of the mobile phase is held constant during the gradient, again achieved through the use of a ternary gradient in which the third eluent component (Mobile Phase C) contains not only the buffer but also TFA in the correct proportions to achieve the desired (constant) mobile phase pH and ionic strength during the gradient. In this way, any selectivity differences due to the analyte/stationary interactions are more easily interpreted compared to the situation in which both eluent pH and ionic strength vary over the course of the gradient.
However, the advantages gained using this approach are not limited to the method development laboratory or column screening procedures. Methods designed with this approach in mind tend to be more robust and have flatter, less noisy baselines. This is illustrated in Figure 3, which shows the use of balanced buffer approach rather than a ternary gradient as described previously. Further, rather than simply matching the additive concentration in both eluent components (for example, 0.1% TFA in both aqueous and organic), the user has balanced the UV absorbance between the phases and adjusted the concentrations according – for example, 0.1% TFA (aq) and 0.03% TFA in Acetonitrile produce the same UV response. In this the flattest possible baselines are ensured during the gradient.

Figure 3: Figure 4(a) gradient baseline shift observed during method development of a stability-indicating method for an NCE using mobile phase A (0.1% trifluoroacetic acid) in water and mobile phase B (0.1% trifluoroacetic acid in acetonitrile) (~0.1 AU at 230 nm) b) reduction of the gradient shift to 0.002 AU at 230 nm achieved by adjusting the concentration of trifluoroacetic acid in mobile phase B to 0.03%, which has a similar apparent absorbance at 230 nm with 0.05% trifluoroacetic acid in water. Reproduced from Reference 1.
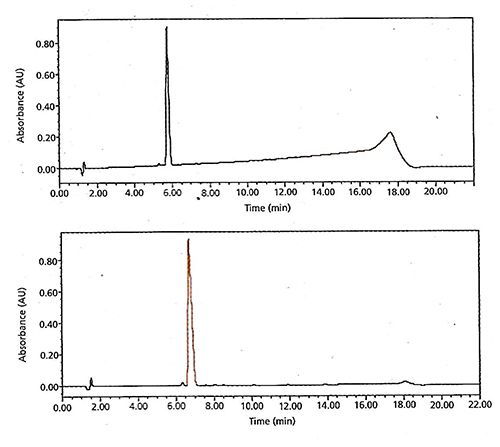
Of course, one should always bear in mind that at higher UV wavelengths, baseline disturbances due to additives such as TFA and other volatile additives have a lessened effect as their absorbance reduces and effects on the refractive index of the solvent become less significant. Further, one might consider the use of a reference wavelength to assist in overcoming baseline drift effects when using a diode array detector. Further information on these topics can be found in Reference 2.
When using TFA as a mobile-phase additive, and working at relatively low wavelengths in order to obtain the required sensitivity for non-aromatic or non-conjugated analytes, we are able to take advantage of the rather peculiar absorbance characteristics of TFA, which are outlined in Figure 4.
Figure 4: Baselines for trifluoroacetic acid–acetonitrile gradients of 0–100% B in 100 min. A: 0.1% trifluoroacetic acid in water; B: 0.1% trifluoroacetic acid in acetonitrile. Absorbance scale is relative, not absolute. Adapted from Reference 2. Reproduced from Reference 2.
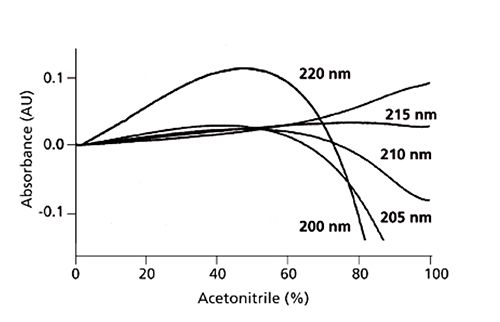
One can see from Figure 4 that at 215nm, as opposed to slightly higher or lower wavelengths, TFA shows a particularly flat response in terms of changing absorbance with increasing acetonitrile. Therefore working at 215 nm may present a significant advantage in terms of reduction of baseline drift during gradient analysis.
References
- M.W. Dong and B.E. Boyes, LCGC Europe,31(10), 572–583 (2018).
- J.W. Dolan, LCGC North America,31(7), 538–543 (2013).


Tony Taylor is the technical director of Crawford Scientific and ChromAcademy. He comes from a pharmaceutical background and has many years research and development experience in small molecule analysis and bioanalysis using LC, GC, and hyphenated MS techniques. Taylor is actively involved in method development within the analytical services laboratory at Crawford Scientific and continues to research in LC-MS and GC-MS methods for structural characterization. As the technical director of the CHROMacademy, Taylor has spent the past 12 years as a trainer and developing online education materials in analytical chemistry techniques.
. From industry he went on to Florida State University, where he was assistant professor of both analytical and materials chemistry. Since 2011, he has been at the National Institute of Standards and Technology (NIST), where he is currently Scientific Advisor in the Chemical Sciences Division. André is the author of over 90 peer-reviewed scientific publications, lead author of the second edition of “Modern size-exclusion liquid chromatography,” editor of the book “Multiple detection in size-exclusion chromatography,” past associate editor of the Encyclopedia of Analytical Chemistry and, since 2015, editor of Chromatographia. He has received a number of awards, including the inaugural ACS-DAC Award for Young Investigators in Separation Science, and was also inaugural Professor in Residence for Preservation Research and Testing at the US Library of Congress. His interests lie principally in the area of macromolecular separations, both fundamental and applied.")



.")


