The LCGC Blog: Back to Basics: The Role of Thermodynamics in Chromatographic Separations
It's important for chromatographers (particularly those who are also graduate students) to understand the fundamental aspects of separation science. In this installment of the LCGC Blog, Kevin Schug discusses the connection between thermodynamics and chromatography.
The spring semester always brings with it a plethora of graduate student defense proposals and updates here at UT Arlington. And, given the number of faculty members in our department who focus their research on aspects of separation science, some of the questioning inevitably turns to fundamental aspects of chromatographic separations. If a student recalls this theory (they should have been exposed to these fundamentals both in undergraduate instrumental analysis, or its equivalent, and a graduate class in separation science by this time), then it is a pretty straightforward line of questioning to get through. However, given that many of today’s research projects feature large collaborative efforts or cut across the boundaries of different topics or disciplines, sometimes thoughts about what’s happening at the molecular level inside a chromatographic column get lost in the bigger picture. I remember being caught off guard by a question during my defense that I was not completely prepared to answer. My PhD mentor (happy 80th birthday by the way, Prof. McNair) asked me to describe the mechanism of how a flame ionization detector works — apparently people still debate this, so I was really not expected to know the answer — I didn’t. It is one of those times that you either pull yourself together and come up with a plausible response, or the nerves get the best of you, and your mind locks up like a steel trap that will not release what it has inside. During a recent presentation by a student, the questioning delved into the connections between chromatographic separations and basic thermodynamics; it was not the best outcome for the student, so I thought I would take the opportunity here to lay out some of the basics that pertain to this topic, in hopes of helping some future students avoid a similarly uncomfortable experience.
Chromatography is a dynamic process involving the repeated transfer of analytes between a flowing mobile phase and a fixed stationary phase. The physicochemical properties of the analyte and the different phases control the degree to which this partitioning happens. Analytes that interact strongly with the stationary phase will be highly retained, whereas those that do not will be rapidly swept through the chromatographic system with the mobile phase. Although equilibrium is never achieved, thermodynamic equilibrium concepts can be applied to better characterize or understand the molecular-scale energetics (that is, free energy, enthalpy, and entropy) of the system.
The simplest connection between thermodynamics and chromatography can be made by the relationship between a capacity (or retention) factor (k’) and an equilibrium constant (K) for an analyte. These are related as shown in equation 1 by the phase ratio (β), which is the ratio of the volume of mobile phase to the volume of stationary phase in the chromatographic system:
K = k’ β [1]
The equilibrium constant, for an analyte that is partitioning from the mobile phase into the stationary phase, is the ratio of the equilibrium concentration of the analyte in the stationary phase to that of analyte in the mobile phase. The capacity factor is the ratio of the adjusted retention time (t’R = tR – t0) to the dead time; it can also be thought of as the amount of time the analyte spends in the stationary phase (t’R) divided by the amount of time the analyte spends in the mobile phase (t0; all analytes spend t0 amount of time in the mobile phase). These (as well as β), are all unitless quantities, but if you think about taking a time average, then the value of the capacity factor should be representative of the ratio of the amount of analyte in each phase at any given point in time, and then multiplication by the phase ratio to obtain the equilibrium constant makes sense.
The equilibrium constant can of course be related to the Gibbs free energy change (ΔG), by the well-known equation,
ΔG = -RT lnK [2]
where R is the gas constant and T is temperature (in Kelvin). The Gibbs free energy change is related to the change in enthalpy (ΔH) and change in entropy (ΔS) for a system by the Gibbs-Helmholtz equation:
ΔG = ΔH – TΔS [3]
A spontaneous “reaction” (in this case, speaking about the analyte favoring interaction with the stationary phase) occurs when ΔG is negative (that is, processes that give up free energy are favorable). This is the same thing as saying that K > 1 (according to equation 2). A spontaneous process could be facilitated by a decrease in enthalpy (that is, ΔH is negative) or by a large increase in entropy (that is, even if ΔH is positive, a positive ΔS, multiplied by the temperature, could also result in an overall negative ΔG and a favorable process).
So, why does this matter? The mode, or the types of noncovalent forces, by which analytes interact with the stationary phase controls the role of enthalpy and entropy in the phase transfer, or partitioning, process. Without going into a great amount of detail, interactions found in reversed-phase chromatography are typically driven by the hydrophobic effect. We also say that such interactions are entropically driven; the system gains entropy when the solvophobic nature of the analyte facilitates its leaving of the mobile phase to interact with the stationary phase. Conversely, electrostatic and dipole–dipole (or hydrogen bonding) interactions are generally enthalpically driven; these are favorable because the interaction between functional groups on an analyte and those on a stationary phase (think ion exchange, hydrophilic interaction, or some modes of chiral chromatography) releases enthalpy in the system. Even though the system is more “ordered,” which is not favorable, the decrease in enthalpy causes the interaction to be spontaneous overall (ΔG is negative). Creating chromatographic separations where the interactions (or the energetics of partitioning) are consistent from one run to the next is important. This is the basis for reproducibility in chromatographic separations; it is also why one must typically reequilibrate the column between gradient analytical runs — to make sure that the chromatographic phases have the same physicochemical properties each time a separation is run.
Another important consideration in separations is selectivity (α). This can also be related to fundamental thermodynamic concepts through similar routes as those presented above. Each analyte has its own capacity factor (as each has its own equilibrium constant, according to equation 1), and we know that selectivity is given by the ratio of capacity factors for these analytes (denoted analyte 1 and analyte 2 here):


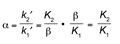
[4]
Thus, we can show that selectivity is achieved when the change in Gibbs free energy for two analytes in a chromatographic system is different; in other words, ΔG2 – ΔG1 = ΔΔG ≠ 0. The difference in free energy change between analyte 1 and analyte 2 can be directly tied to selectivity through the following equations:
ΔG2 = -RT lnK2
ΔG1 = -RT lnK1
ΔG2 – ΔG1 = ΔΔG = -RT lnK2– (-RT lnK1)
= -RT (lnK2 – lnK1)
= -RT ln (K2/K1)
ΔΔG = -RT lnα [5]
Thus, you could even scrutinize the energetics (in terms of enthalpy and entropy) leading to favorable selectivity between two analytes. This type of analysis is actually quite important in chiral separations, where often very small differences in free energy change for the interaction of analyte enantiomers with a chiral stationary phase need to be optimized in order attain separation. Of course, selectivity is only part of the equation (literally!). To separate compounds, we need to resolve them (think “resolution”), and this is a function of not only thermodynamic (capacity factor and selectivity), but also kinetic processes. The efficiency of the separation, which is often described by numbers of theoretical plates, is largely controlled by band broadening parameters, which are more time-dependent (that is, kinetic). But, this is a discussion for another time. For now, at least, if you are one of those students who gets asked how chromatographic concepts relate to fundamental thermodynamics, you have no excuse if you do not impress your evaluation committee with your understanding of this topic.
Previous blog entries from Kevin Schug:
The LCGC Blog: The Dimensionality of Separations: Mass Spectrometry Is Separation Science
The LCGC Blog: What Can Analytical Chemists Do for Chemical Oceanographers, and Vice Versa?
Restricted-Access Media for Biomonitoring Applications: A Solution That Deserves More Attention
.")


The LCGC Blog: Historical (Analytical) Chemistry Landmarks
November 1st 2024The American Chemical Society’s National Historic Chemical Landmarks program highlights sites and people that are important to the field of chemistry. How are analytical chemistry and separation science recognized within this program?


.")

