The LCGC Blog: Automated Dynamic Liquid-Liquid Microextraction (DLLME) – Improve Sample Extraction, Buy Some Time, and Reduce Environmental Footprint
The improved performance of chromatographic detectors, most notably mass spectrometers, has enabled many advances in analytical science, however, one such advance may be given less prominence than perhaps it should.
The improved performance of chromatographic detectors, most notably mass spectrometers, has enabled many advances in analytical science, however, one such advance may be given less prominence than perhaps it should.
Typically, the volumes of liquid handled by automation “robots” are limited. Depending on the sample preparation application, this limitation in sample volume, alongside speed with liquids, can be handled. The achievable sample enrichment factors mean that smaller-scale liquid handling robots have been excluded from many important areas of analysis.
That has now changed significantly. We need to consider the positive implications that this may have on the quality, safety, and efficacy of our analytical measurement.
In the past decade, the sensitivity of mass spectrometric detectors (for example, triple quadrupole, QqQ systems) has improved vastly, and an order of magnitude or greater increase in sensitivity has been achieved. Instrument manufacturers demonstrate sub femtogram instrument detection limits (IDL) for GC–MS systems and even single quadrupole GC–MS instruments are now capable of IDLs in the single-digit femtogram range. Triple quadrupole detectors used with HPLC that have electrospray atmospheric pressure ionization sources are stated to have IDLs in sub 100 femtogram with several claiming limits of half as much.
How does this enable the use of fully automated, online sample preparation systems to perform trace analysis?
The sample volume for typical, online sample handling robotics is usually less than 50 mL, due to the way in which syringes or low-volume pipettes are used for sample transfer and manipulation. This affects the speed with which this can be achieved. The instrument needs to achieve either a very high sample enrichment factor or very low detection limits. The combination of the improvement in mass spectrometric detector sensitivity with the introduction of extraction techniques-primarily the so-called “microextraction” techniques-means that sample preparation is now possible using automated online systems. Previously, this could only be achieved using traditional liquid–liquid or large volume solid-phase extraction.
So why is this important to us?
Anyone who has labored with manual sample extraction using shake flask techniques will know that these are fraught with issues, including the following:
- Large volumes of organic solvent are typically required, which is both environmentally damaging and a health hazard to the operator
- Attendant dangers of flask droppage or spillage
- Manual shaking or even shaking with an orbital mixer produces variable results
The extracting solution needs to be pre-concentrated prior to injection, aand this stage is highly prone to contamination and irreproducibility.
Automated, online sample preparation is capable of overcoming all of these disadvantages and achieving very low detection limits with only limited sample volumes. When operated in parallel processing mode, it can reduce the required sample preparation time to below the duty cycle time of the chromatographic system-improving throughput alongside the benefits of improved reproducibility. The benefits of improved accuracy, reproducibility, and reduced contamination are often associated with automated sample preparation.
Due to the increased detector selectivity associated with MS detection, where analyte signals may be selectively extracted using mass filtering, a general move towards simplification of sample preparation operations has been found. The number of applications published which use simple liquid–liquid extraction has markedly increased.
One such microextraction technique which is rapidly gaining in popularity is dynamic liquid–liquid microextraction (DLLME). The principle of the technique (which is distinct from traditional liquid–liquid extraction approaches), the facile sample processing paradigm and the extraction and enrichment benefits are very impressive.
DLLME uses a small volume of immiscible extracting solvent to form an emulsion of micro-droplets within the sample. This presents a very high surface area for analytes to partition into. In order to increase the “contact” between the two phases, a disperser solvent is used which is highly miscible with both phases and serves to reduce the interfacial tension between the sample and extracting solvent. After addition of the extracting and disperser solvents, the sample is vigorously mixed or vortexed to affect extraction. Importantly, due to the very high surface area contact, the time taken to reach equilibrium (maximum extraction) is relatively short. Following extraction, the sample is vortexed and the extracting solvent aspirated for subsequent analysis.
DLLME is used with aqueous samples and an organic extracting solvent. However, it is also possible with a slight adaptation to the approach, to extract analytes from organic samples with aqueous solvents. Crucially, the technique can achieve high enrichment factors as only a relatively small volume of extracting solvent is used and an increase in analyte concentration of 10–50x is typical.
A schematic representation of the DLLME process is shown in Figure 1:


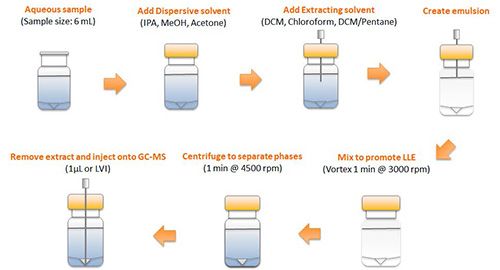
Figure 1:Schematic representation of the DLLME process
Implementation of automated online DLLME has required the development of several novel robotic tools which are worthy of further note. In order to rapidly transfer fairly large amounts of sample, pipetting tools are required to achieve sample transfer in reasonable timeframes. Vortex mixing is crucial in order to affect speedy and reproducible extractions, and therefore, an automated vial vortexing tool is required. The development of a solvent-safe automated centrifuge is required in order to break the emulsion and form extractant sediment within the well of a high-recovery vial (where the extractant is denser than the sample). The final injection solution is aspirated from here.
Typically, the automated DLLME process can be carried out in a comparable timescale to the chromatographic run-time. With batch preparation or parallel processing modes, the sample preparation time for multiple samples is considerably shorter than the combined chromatographic analysis time for all samples.
This high-speed aspect is also important when considering method development and optimization. The factors which are typically considered in this stage might include:
- The volume of Sample (1–30 mL*)
- Nature** of volume of extractant (100–500 μL)
- Nature*** and volume of disperser (5–10x extracting solvent volume)
- Sample pH adjustment (to promote extraction from aqueous to organic solvents, pH may be adjusted to reduce the degree of ionisation of target analytes)
- Effects of salt addition (increasing the electrolytic strength of an aqueous sample can help promote the partition of polar analytes from an aqueous phase into an organic extractant)
- Vortex or mixing (extraction) speed and time (typically a few s to a few min)
- Injection volume (note that Large Volume Injection GC can be used to improve detection limits even further with most modern GC system having the ability to injection up to 1000 μL of the sample)
Notes:
* The volumes shown in the table above are typical but will vary depending upon the sample type, desired degree of enrichment etc.
** The full range of aqueous immiscible organic solvents are available to use as the extracting solution
*** Typical disperser solvents include isopropanol, acetone, methanol, acetonitrile, and dichlorobenzene
Due to the very fast sample extraction, it is possible to optimize the DLLME procedure using a one-factor-at-a-time (OFAT) approach. Several publications cite the use of this experimental design to more efficiently arrive at the optimum experimental conditions. A simple example is shown in Figure 2 below:

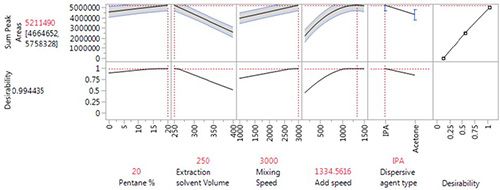
Figure 2:Typical responses from a simple Design of Experiments (DoE) approach to DLLME optimization
Alongside the parameters considered in Figure 2, length of extraction time was not found to be a significant factor in the range 0.6 s to 5 min, indicating, for this application, the high speed, and the efficiency of the DLLME technique.
Figure 3 shows the MRM chromatogram and calibration curve produced following DLLME sample preparation of Aldrin as part of a suite analysis of organochlorine pesticides (OCP) and polychrorinated biphenyls (PCB) from surface water, for which the figures of merit achieved are also shown:

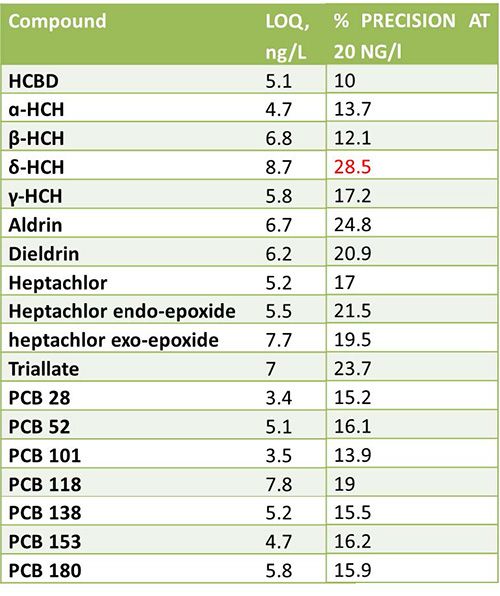

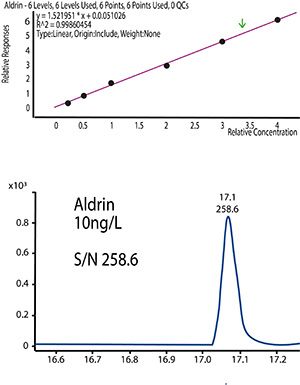
Figure 3:Results from the validation of OCP and PCB DLLME sample extraction and subsequent analysis with GC–MS/MS
Despite the judicious choice of extracting solvent, sample pH and electrolytic strength optimization; the fact remains that liquid–liquid extraction is not a highly selective sample preparation technique. However, as mentioned above, the use of the more highly selective triple quadrupole mass spectrometric detectors can help to mitigate this factor. It is also possible to combine the use of DLLME with more highly selective μSPE or molecularly imprinted sorbent (MIP) extraction, where the benefits of semi-targeted sample enrichment and the more highly selective micro-sorbent extraction can also be combined in a rapid and automated fashion to produce highly selective and concentrated sample extracts. This may be an advantage when using LC–MS/MS techniques.
The DLLME process is versatile and can be applied to any situation in which liquid–liquid extraction is typically used. It can transform a manual and time-consuming sample preparation method into a rapid, sensitive, and fully-automated online technique. To help justify the rather bold statements made in the title of this piece, Figure 4 shows a graphical representation of some proposed advantages of the DLLME technique for an application based on a manual liquid–liquid extraction using 50 mL of DCM and 400 μL for the automated method:

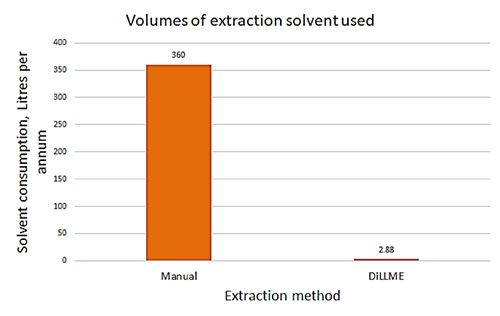

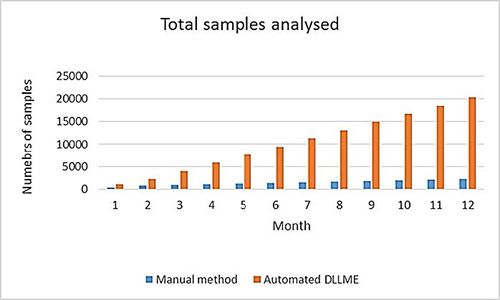
Figure 4: Comparison of manual liquid–liquid extraction and automated DLLME approaches to sample extraction
Automated DLLME is a technique which can exploit the improvements in mass spectrometric sensitivity and micro-extraction techniques to deliver benefits in speed, quality, and efficacy of sample extraction and enrichment. Significant solvent reduction, in comparison to traditional approaches, can positively impact environmental footprint.


Tony Taylor is the technical director of Crawford Scientific and ChromAcademy. He comes from a pharmaceutical background and has many years research and development experience in small molecule analysis and bioanalysis using LC, GC, and hyphenated MS techniques. Taylor is actively involved in method development within the analytical services laboratory at Crawford Scientific and continues to research in LC-MS and GC-MS methods for structural characterization. As the technical director of the CHROMacademy, Taylor has spent the past 12 years as a trainer and developing online education materials in analytical chemistry techniques.



.")


The LCGC Blog: Historical (Analytical) Chemistry Landmarks
November 1st 2024The American Chemical Society’s National Historic Chemical Landmarks program highlights sites and people that are important to the field of chemistry. How are analytical chemistry and separation science recognized within this program?

