The LCGC Blog: Are we scared to properly explore selectivity options in HPLC?
Much has been written about options for increasing efficiency in HPLC ? primarily through the use of core shell and sub 2 ?m particles, which have been used to increase efficiency, speed up separations or increase peak capacity. However, many separations can only be effectively improved, by optimising selectivity ? resolution is a function of selectivity, efficiency and retention, with selectivity being the most effective at achieving good resolution.
Are we scared to properly explore selectivity options in HPLC?
Much has been written about options for increasing efficiency in HPLC – primarily through the use of core shell and sub 2 μm particles, which have been used to increase efficiency, speed up separations or increase peak capacity.
However, many separations can only be effectively improved, by optimising selectivity – resolution is a function of selectivity, efficiency and retention, with selectivity being the most effective at achieving good resolution.
Optimizing separations through improved selectivity has become less fashionable, or perhaps more daunting and time consuming. In evidence, I offer the proliferation of methods which use 0.1% TFA as a"'buffer" (which it isn't) to keep well away from analyte pKa values, when using a highly hydrophobic phase such as C18 and acetonitrile as the organic modifier to control retention (and separation). This has almost become the "de facto" template for modern HPLC methods – especially when MS detection is used.
At best, the most advanced labs will have a screening platform with a selection of "orthogonal" chemistries which are capable of various pH/organic modifier combinations. This is perhaps utilised in combination with computer aided optimisation software or as part of statistical experimental design. We hope that this approach follows Pareto's Law (which states that for many events, roughly 80% of the effects come from 20% of the causes) and for the 20% of cases where a method does not 'drop out' of the screen, then we have to do some "real work."
There is absolutely nothing wrong with this approach – other than it does not encourage thought about the separation process and the nature of the interaction between the analytes, stationary phase chemistry and mobile phase. Such thought is often required in order to achieve suitable separations for complex samples, analytes which are chemically very similar or for bio-separations in which we are investigating small chemical differences within a large protein molecule.
I often give lectures on column selectivity and the effect of various stationary phase characteristics and these talks often feature a section on mixed mode and hydrophilic interaction chromatography (HILIC) which have emerged as useful alternatives to the standard reversed phase mode. However, one ever present question after such lectures is on the complexity of these separations and the number of interactions which need to be understood in order to properly understand the retention mechanisms and build a robust, optimised separation. Well, robust and high throughput is fine – but difficult separations often need to be solved with specialist approaches and sometimes 'shock horror' we even need to think about the nature of the separations at hand.
I'll quote two very simple examples which I hope will prove how useful the more complex of these approaches, mixed mode, can be.
Glycans are sugar moieties which, when bound to proteins such as monoclonal anti-bodies, can be used in recognition and regulatory processes – a little like the zip code to our proteins "envelope." Adjusting the glycan moiety can be used to fine tune the protein function, efficacy and safety, and regulators require a detailed characterisation of this moiety during development and manufacture. Glycans are highly polar, take many different forms and can be differentiated by hydrophobicity, size (volume) in solution, position and type of substituent sugars and charge.
Traditional techniques such as reversed phase ion pair chromatography and anion exchange chromatography were used for the analysis of glycans – but both of these techniques are unfashionable and difficult to interface with mass spectrometry. HILIC chromatography can be used to distinguish differences in size and composition (hydrophobicity), but cannot be used to differentiate based on charge state.
Mixed mode chromatography, using ligands which have hydrophobic and electrostatic moieties (anionic, cationic or zwitterioninc, Figure 1), can separate glycans on the basis of size, composition and charge, Figure 2.


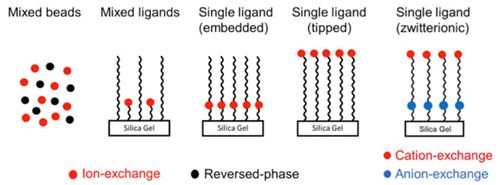
Figure 1: Various methods of producing mixed mode HPLC stationary phases

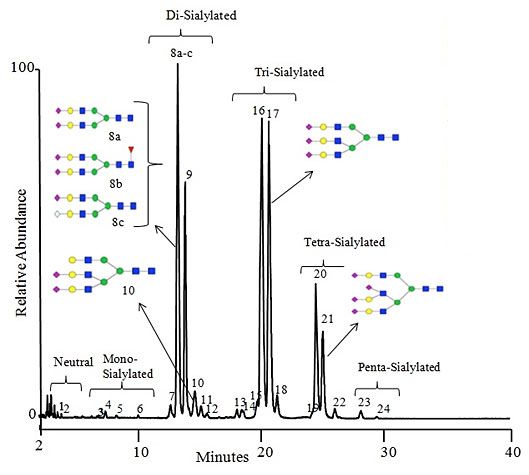
Figure 2: Separation of underivatised glycans from bovine fetuin using an anion exchange mixed mode HPLC column
GlycanPac AXH-1 column (1.9 μm, 2.1 × 150 mm)
A: Acetonitrile:water (80:20), B: Ammonium formate (80mM pH 4.4)
2.5% to 37.5% B in 40 mins. linear gradient, 0.4mL/min, 30oC, 200 pmoles on column, MS detection, negative ion mode - Reproduced with permission of ThermoFisher Scientific
Even though these separations are complex, there are 'screening' methods which explore the selectivity differences obtained by altering the eluotropic strength, eluent pH and the nature or concentration of the buffer, which are the three common factors, used to optimise the selectivity of mixed mode separations. Even though more thought needs to go into these separations – there are generic approaches to exploring the range of separation possibilities.
These generic approaches can involve TFA based separations; therefore, our generic screening can result in methods which are suitable for MS detection. Figure 3 shows the range of selectivity which can be explored for a sample containing acidic, basic and neutral analytes by adjusting pH and elutropic strength using a zwitterionic stationary phase.

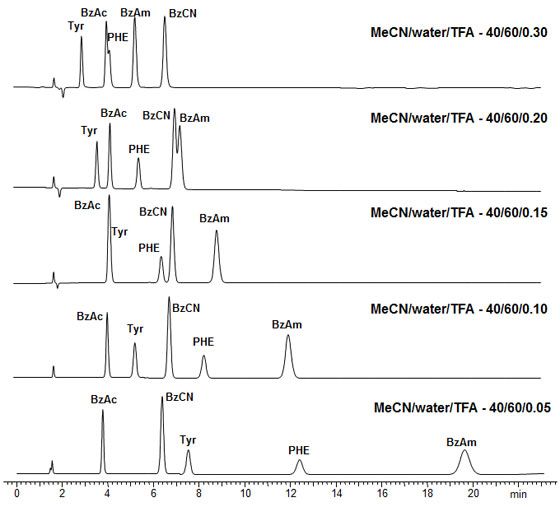
BzAc - Benzoic acid, BzCN - Benzonitrile, TYR - Tyrosine, PHE - Phenylalanine, BzAm- Benzylamine
Obelisc R (Zwitterionic), 150 x 4.6 mm, 1.0 mL/min, UV 250 nm, Mobile phases as shown

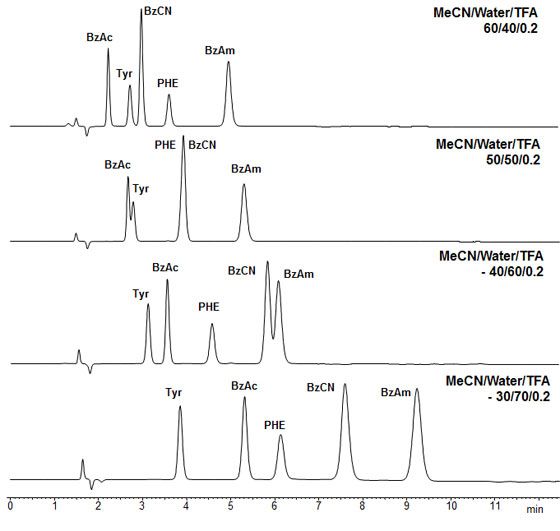
Ac - Benzoic acid, BzCN - Benzonitrile, TYR - Tyrosine, PHE - Phenylalanine, BzAm- Benzylamine
Obelisc R (Zwitterionic), 150 x 4.6 mm, 1.0 mL/min, UV 250 nm, Mobile phases as shown
Figure 3: Separation of acidic, basic a neutral analytes using a simple screening approach
with a zwitterionic mixed mode stationary phase - Reproduced with permission from SIELC Technologies
It is true that in order to understand and properly optimise a separation, one often needs to understand the chemistry of the analytes and the stationary phase, for example to know if the electrostatic groups on the stationary phase are ionised or non-ionised at various eluent pH values.
It is also true that these columns often need more time to equilibrate initially.
However, just because the columns can't be abused and because we need to think about the underlying chemistry of the separation, this doesn't make them unsuitable for us to use.
The separation of highly polar analytes, analytes with very similar hydrophobicity, mixtures of acids bases and neutrals and even the separation of analytes and their counter ions can be successfully achieved using this type of stationary phase. So why don't I see them used in every laboratory? Well, at least in part, in think it's because selectivity optimisation is less fashionable, a little bit more difficult and often too much like hard work!



.")


The LCGC Blog: Historical (Analytical) Chemistry Landmarks
November 1st 2024The American Chemical Society’s National Historic Chemical Landmarks program highlights sites and people that are important to the field of chemistry. How are analytical chemistry and separation science recognized within this program?

