The LCGC Blog: Ammonium Acetate Woes
I’ve been dealing recently with issues in the laboratory when using ammonium acetate buffers, including surprising rises in HPLC–MS back pressures when starting the instrument after overnight storage, as well as difficulties with MS sensitivity.
I’ve been dealing recently with issues in the laboratory when using ammonium acetate buffers, including surprising rises in HPLC–MS back pressures when starting the instrument after overnight storage, as well as difficulties with MS sensitivity.
I also frequently see posts on chromatography forums relating to increasing instrument back pressure when using ammonium acetate (and indeed ammonium formate), especially after standing overnight or with the first few runs of each day. Much head scratching can result from the use of high-soluble aqueous buffers which become problematic in practical use, with blocked capillaries, pre-columns, and analytical column end frits.
These problems invariably result from misunderstandings of buffer solubility in mixed organic aqueous media, which can be overcome with the data shown in Figure 1.


Figure 1: Solubility of five buffers in mixtures with acetonitrile where - represents ammonium acetate at pH 5.0, •••• represents ammonium phosphate at pH 3.0, --- represents potassium phosphate at pH 3.0, –••– represents ammonium phosphate at pH 7.0, and – – represents potassium phosphate at pH 7.0. (Adapted with permission from Ref. 1.)
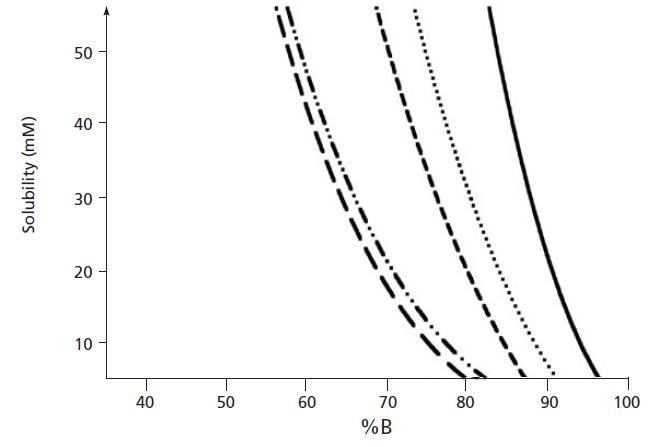
From Figure 1, it can be clearly seen that the solubility of ammonium acetate in binary mixtures containing above 90% acetonitrile is increasingly limited in solubility, and is wholly insoluble in 100% acetonitrile. While 20 mM ammonium acetate is the solubility limit at 90% acetonitrile, this limit falls quickly to 10 mM ammonium acetate (a popular buffer concentration used with LC–MS applications) at 95% acetonitrile mixtures. If these solubility limits are exceeded, then a cloudy liquid will result, due to the fine ammonium acetate precipitate within the solution, which of course can block capillaries and column frits, causing increases in system back pressure. It should also be noted that the data from Figure 1 are derived using a high-quality buffer. Solubility will decrease if lower-quality (purity) buffer salt reagents are used. Of course, one should never try to dissolve ammonium acetate in pure acetonitrile, and even though initially it may appear that the buffer is soluble, it will quickly drop out of solution (Figure 2).

Figure 2: Ammonium acetate residue created when “dissolving” ammonium acetate in acetonitrile, followed by the addition of water to achieve the required organic/aqueous ratio of the eluent. (Photograph courtesy of Dr. Paul Ferguson, Astra Zeneca, UK.)

In a typical reversed-phase HPLC gradient, this is perhaps not so much of a problem, unless of course your gradient goes to >90% acetonitrile. However, there are other considerations, such as the relative percentages of organic solvent the aqueous buffer may encounter when mixing using low or high pressure mixing systems, when injecting samples dissolved in high percentages of acetonitrile or, in the worst of all cases, when systems are flushed with 100% acetonitrile to remove column contaminants or for overnight storage.
There is the further consideration, when using 100% acetonitrile for column storage, that large pH shifts can occur in 100% organic solvent, and one needs to be mindful if these pH shifts might take the column storage pH into a range where the dissolution of the silica matrix may be possible. This would lead to the formation of “fines” within the column, which would ultimately result in column blockages (at high pH), or to the stripping of the bonded-phase ligands (at low pH).
Of course, many of us choose to use ammonium acetate as a buffer, especially when using MS detection due to its inherent volatility and low propensity for API source contamination, however, we need to be very mindful of this limited solubility and adjust our HPLC practice accordingly. On this point, I often ask students and co-workers if the use of ammonium acetate is appropriate for the experiment and indeed, is a buffer necessary at all? There is much misunderstanding relating the requirements for use as well as the appropriate choice and concentration of a buffer, and we will use ammonium acetate as the case in point to investigate the uses and misuses of this much-loved buffer system.
Buffers are required to resist small changes in pH (primarily of the eluent), and to ensure that the HPLC column remains in a state of constant charge (primarily here we consider the state of ionization of residual silanol species on the surface of the silica support). Changes in pH may cause problems with retention time stability, peak shape and , when using Electrospray MS, instrument sensitivity. Typically the biggest ‘challenge’ to the system pH will be on mixing of the sample diluent with the eluent in the connective components between the injector and the HPLC column (or pre-column) and at the head of the column. If the sample diluent has a different pH then the analyte (or a portion of all analyte molecules) may change ionization state and therefore chromatograph differently, or respond differently in the MS interface. However, we need to properly understand the chemistry of our analytical method to make critical choices on the type and concentration of buffer. As we will see, the analyte concentrations and amount of column surface that we need to control are low enough to need only a very low concentration of buffer to maintain reproducible retention times and acceptable peak shapes.
Figure 3 can help us to understand the situations in which ammonium acetate can be of use both chromatographically and mass spectrometrically.

Figure 3: Variation of buffer capacity for aqueous ammonium acetate solution (10 mM) with various proportions of acetonitrile (%) (Adapted from Ref. 2 with permission.)
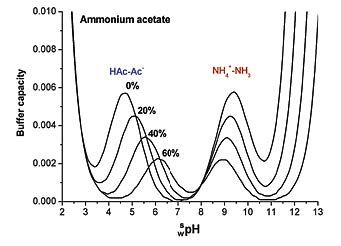
Essentially, there are two buffering regions when 10 mM ammonium acetate is added to our eluent solution, and either dilute ammonia or formic acid are used to adjust the pH (note that without the addition of the acid or base, the solution will have very little buffering capacity).
In 100% aqueous solution, the buffer pKa values around 4.8 and 9.5, and from our basic chromatography lessons we know that the buffer is best used around +/- 1 pH unit from the buffer pKa where the buffering capacity will be reduced to around 66%, and at 2 pH units away it is around 5%. So, for our ammonium acetate buffer in water, the eluent pH used for our separation should be 3.8 to 5.8 when using formic acid as the pH modifier, and 8.5 to 10.5 when ammonia is used to adjust the eluent pH. However, the caveat here is that once acetonitrile is added to the system, this working range changes, and the usable pH range becomes 5.2 to 7.2 or 7.9 to 9.9 once 60% acetonitrile has been added. Of course, for gradient separations the pKa of the buffer system will change constantly and we recommend that the pKa of the system at the starting gradient composition is used to estimate the usefulness of the buffer for the separation.
So, the key question is, will these pH ranges be sufficient to avoid changes to the extent of analyte ionization or column protonation changes? For MS detection we typically choose to the have our ionogenic analytes in the ionized form for good detection sensitivity, managing reversed-phase retention through judicious choice of stationary-phase and organic solvent type and composition. So, if our analyte is fully ionized at the pH ranges suggested above and in Figure 3, then we should be fine, with one caveat. We should also note from Figure 1 that the buffer capacity of the system reduces as acetonitrile is added, with the buffer capacity dropping to 30% of the aqueous value at 60% acetonitrile. The key principle with buffer use in LC–MS is to use as little as possible to maintain retention time reproducibility, acceptable peak shape, and detector sensitivity, as the concentration of buffer will directly influence the amount of ion suppression encountered, and therefore method sensitivity will be directly affected. To maintain good buffering capacity at lower buffer concentrations, we often aim to work within +/- 0.5 units of the buffer pKa.
Table I therefore shows the recommended pH ranges in which ammonium acetate buffers will be useful.

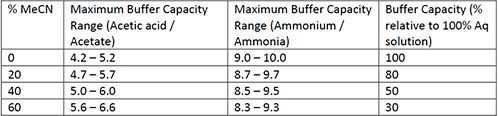
From Table I we can use the buffering ranges to select the eluent pH in which our analyte should be 100% ionized, noting that the buffer concentration used to derive these figures is 0.1 mM, a popular choice for buffer concentration when using MS detection. Typically for basic analytes, the acetic acid/acetate buffering system is used, and the eluent pH is usually well below the pKa of the basic analytes, which ensures that they are constantly protonated. The same may be true of acidic analytes with the ammonium/ammonia system, where acidic analytes should all be fully deprotonoated. This will ensure that retention times are steady, peak shapes are good, and the MS sensitivity is optimized from a method chemistry perspective.
It should be immediately obvious that there are large gaps in the effective buffering ranges for ammonium acetate-based eluents. That is, at a 20% acetrontrile eluent composition (or starting gradient composition of 20% acetonitrile), there is likely to be lower buffering capacity (higher buffer concentrations will need to be used) with an eluent pH below 4.2, between pH 5.2 and 9.0, or above 10.0. I’m sure there will be methods out there where eluent pH is adjusted outside these recommended ranges when using ammonium acetate buffers, and one may need to consider using a different buffer system, and the formate/formic acid system is a popular choice at eluent pH values below 4.2.
One final point is worthy of note here. If our eluent pH is far away from the pKa of the analyte, small changes in eluent pH will have little effect on the degree of analyte ionization, and under these circumstances the use of a buffer may be unnecessary. For example, acetic as well as formic, trifluoro and difluoroacetic acids have a substantial ‘self-buffering’ capacity a low pH, and provided the basic analyte pKa >2 pH units higher and acidic analyte pKa <2 pH units lower than the eluent pH, then the use of a buffer salt is often unnecessary.
Of course, here I’m assuming that basic analytes are analysed using an acidic eluent system and acidic analytes with a more basic eluent system pH to ensure full ionization and good electrospray MS sensitivity. So, if you are experiencing instrument issues with instrument back pressure, retention time instability, or MS detection sensitivity, then it may well be worth considering if a buffer salt is required at all, and that the judicious use of formic or acetic acids or ammonia solution may well solve your issues.
Whatever the case, you must take time to understand the chemistry of the method with respect to your analyte pKa, the required eluent pH, and the choice of buffering system used to attain and maintain this pH within the system.
For those who do not have analyte pKa information, there are a number of free programs available which do a reasonable job of predicting analyte pKa based on structure (ChemSketch (https://www.acdlabs.com/resources/freeware/chemsketch/) and MarvinSketch (https://chemaxon.com/products/marvin) are widely used in our laboratory)
For those who seek a deeper understanding on buffers and their use within HPLC and LC-MS I refer you to the excellent articles in references 3-6.
References:
- A.P. Schellinger, P.W. Car, LCGC North America22(6) (2004).
- X.Subirats, E. Bosch, and Marti Rosés, LCGC North America27(11) (2009).
- “Mobile-Phase Buffers, Part I - The Interpretation of pH in Partially Aqueous Mobile Phases,” LCGC North America20(11) (2002).
- “Mobile-Phase Buffers, Part II - Buffer Selection and Capacity,” LCGC North America20(12) (2002).
- “Mobile-Phase Buffers, Part III - Buffer Selection and Capacity,” LCGC North America 21(1) (2003).
- “Mobile-Phase Buffers in LC: Effect of Buffer Preparation Method on Retention Repeatability,” LCGC North America37(7) (2019).


Tony Taylor is Group Technical Director of Crawford Scientific Group and CHROMacademy. His background is in pharmaceutical R&D and polymer chemistry, but he has spent the past 20 years in training and consulting, working with Crawford Scientific Group clients to ensure they attain the very best analytical science possible. He has trained and consulted with thousands of analytical chemists globally and is passionate about professional development in separation science, developing CHROMacademy as a means to provide high-quality online education to analytical chemists. His current research interests include HPLC column selectivity codification, advanced automated sample preparation, and LC–MS and GC–MS for materials characterization, especially in the field of extractables and leachables analysis.
. From industry he went on to Florida State University, where he was assistant professor of both analytical and materials chemistry. Since 2011, he has been at the National Institute of Standards and Technology (NIST), where he is currently Scientific Advisor in the Chemical Sciences Division. André is the author of over 90 peer-reviewed scientific publications, lead author of the second edition of “Modern size-exclusion liquid chromatography,” editor of the book “Multiple detection in size-exclusion chromatography,” past associate editor of the Encyclopedia of Analytical Chemistry and, since 2015, editor of Chromatographia. He has received a number of awards, including the inaugural ACS-DAC Award for Young Investigators in Separation Science, and was also inaugural Professor in Residence for Preservation Research and Testing at the US Library of Congress. His interests lie principally in the area of macromolecular separations, both fundamental and applied.")



.")


