LC–API-MS(/MS) for Biological Samples: Importance of Hydrophobicity, Molecular Weight and Structural Development
The combination of reversed-phase high performance liquid chromatography (RP-HPLC), atmospheric pressure ionization (API), mass spectrometry (MS) and tandem mass spectrometry (MS/MS) is ideal for determining and characterizing analytes in complex biological matrices. This review looks at the importance of parameters such as hydrophobicity, ionization properties, molecular mass and, partially, the molecular structure resulting from applied LC–MS–MS systems in analytical laboratories. The use of these parameters to investigate biomolecules and their unambiguous identification is also described.
During the last two decades the coupling of high-performance liquid chromatography (HPLC) and mass spectrometry (MS) has become one of the most important analytical tools to identify and quantify polar and thermally unstable organic compounds. Atmospheric pressure ionization (API) is very effective for ion transition and LC–MS analysers are regarded as extremely robust "workhorses", which are as stable and efficient as gas chromatography mass spectrometry (GC–MS) systems.
Liquid chromatography atmospheric pressure ionization mass spectrometry (LC–API-MS) methods for analysing single or multiple organic compounds have been widely published in a variety of applications in the last five years. These include bioanalytical,1 food and nutrition science,2,3 forensic,4 industrial,5 medical,6 pharmaceutical7 and environmental applications.8,9
LC–API-MS systems are used to identify and quantify small and large organic molecules. The analytical assessment of substances from biological sources is a flourishing area of research and the identification of proteins and metabolites from biological matrices is becoming particularly important. This "postgenome-era", produces significant amounts of information and can be divided into "proteomics" and "metabolomics". Researchers in these disciplines are interested in characterizing the changes of protein or metabolite levels in individual biological systems in particular situations. Work in both fields requires high analytical standards and, as a result of the complexity of the analysis, is very labour-intensive and time-consuming. Proteomics and metabolomics approaches have to deal with complex matrices and a huge amount of analytes in parallel.
A large number of proteins can be directly identified from biological assays using liquid chromatography electrospray ionization mass spectrometry (LC–ESI-MS).10 A large amount of proteins found in urine,11E. coli12 and blood plasma,13 can be characterized using this method. These proteins are generally analysed by LC–ESI-MS/MS as molecules with an intact primary sequence or — after enzymatic hydrolysis — as smaller peptides. This approach is being increasingly used for routine analysis.
For most of these applications, analytes have to be analysed from highly complex matrices and often a lot of effort is required for sample preparation. Common methods for sample preparation of biological assays involve molecular selections and enrichments, such as as liquid–liquid extraction (LLE), size exclusion chromatography (SEC), affinity chromatography (AC), isoelectric focussing chromatography (IFC) and hydrophilic and hydrophobic solid-phase extraction (SPE).
These selective sample preparations can be coupled with reversed-phase liquid chromatography (RP-LC). The big challenge, however, is to measure these complex assays directly by RP-LC–API-MS, because, as mentioned previously, these sample preparations are expensive and time consuming, the structure and amount of analytes can change and, therefore, more information can get lost.
Direct analysis requires substances to be "viewed" as unaffected by their matrices and to assign results unambiguously. An excellent approach for this requirement comes from LC–API-MS analysis. Combining the two complementary techniques in this type of analysis allows the determination of two independent parameters, namely hydrophobicity (by LC) and molecular weight (by MS). The additional use of tandem mass spectrometry (MS/MS) may result in the (primary) structure of molecules to be determined. Furthermore, the transfer and ionization of molecules from solvent into gas-phase can sometimes be specifically regulated by the ion sources. Finally, three to four (completely) independent parameters can be observed for any detected molecule and so a robust data set can be guaranteed.
DIN-Standards, EU- and other directives14,15 for the analysis of small organic molecules state that "retention time", molecular weight and specific fragments are a necessary prerequisite to identify and quantify substances. Unfortunately, such guidelines are often missing for the characterization of large protein molecules. There are now many LC–MS methods published for the analysis of essential amino acids, peptides, oligopeptides and proteins.1 The mass spectrometric parameters — molecular weight and molecular fragments — are often included in these publications, the value of hydrophobicity, however, is mostly disregarded. This is unfortunate because hydrophobicity can be measured directly via retention time.
A better understanding of hydrophobicity would also help to eliminate avoidable misinterpretations. A typical example is a glycolysated peptide where the mass spectrometric signal shows only the peptide's backbone without the sugar-moiety. The peptide's hydrophobicity, however, would be abated by the polar glycosidic group and it would elute earlier compared with the original peptide (containing no "posttranslational modification").
This article highlights that more attention should be paid to all ascertainable physical parameters of LC–API-MS analysis for the characterization of known and unknown molecules in bioanalysis.
Experimental
Chemicals: Ammonium acetate, sodium hydrogen carbonate, acetic acid, hydrochloric acid and ethanol were purchased from Merck (Darmstadt, Germany). Acetonitrile (AcN) was purchased from Riedel de Haën (Sigma-Aldrich), acetone from J.T. Baker (Deventer, The Netherlands), the amino acid standard AAS 18 came from Sigma-Aldrich (with 18 natural amino acids: Ala, Arg, Asp, Cys, Gln, Glu, Gly, His, Ile, Leu, Lys, Met, Phe, Pro, Ser, Thr, Tyr, Val) and dabsyl chloride (4-[dimethyl amino]azobenzene-4'-sulphonyl chloride) from Pierce (Rockford, Illinois, USA). The reference substances Phe-Gly, Ala-Tyr, Gly-Asn, Pro-Ser, His-Phe, Gly-Glu, Leu-Tyr, Ala-Lys, Phe-Trp and spontaneous urine from standardized mouse samples were provided by the Chair of Molecular Nutrition Unit of the TU München, the standard Val-Val came from Prof. L. Moroder (MPI of Biochemistry) and the substances Phe-Leu and Leu-Leu had been synthesized earlier in-house (Prof. H. Klostermeyer, formerly Chair of Dairy Technology, TU München). Ultra pure water was taken from a Milli-Q water system (Millipore, Eschborn, Germany).
Reference substances were (partly using interim solutions) adjusted to 10 μM (30% AcN) and transferred into the LC system. Substances eluting within the first five minutes were derivatized in an autosampler (AS 3500, Thermo Separation Products, Darmstadt, Germany) and re-analysed with LC–ESI-MS. A dabsylation according to Krause et al.16 was accomplished for the amino acids and biogenous amines. Only the dilution buffer was changed and thus a mixture of 50 mL elution buffer A (see next chapter), 10 mL ethanol, 20 mL acetonitrile and 20 mL acetone was used. Furthermore, 200 μL methanol was added to the reaction vessel after dabsylation. This vessel was put into the autosampler and the sample was re-analysed.
LC–ESI-MS instrumentation and method: The samples were separated using the liquid chromatograph HPLC 1100 micro series (Agilent, Waldbronn, Germany) and analysed using an electrospray-coupled Q-ToF mass spectrometer (Ultima, Waters-Micromass, Manchester, UK). The HPLC system was controlled by ChemStation software (Rev. B01.01, Agilent, Waldbronn, Germany) and the mass spectrometer by MassLynx 4.0 software (Waters-Micromass, Manchester, UK). The data analysis was also performed by the MassLynx 4.0 software.
The chromatographic separation was accomplished by an Agilent Zorbax C18 column (150 × 0.5 mm; 5 μm) with AcN/water at a flow-rate of 20 μL/min and with an injected volume of 8 μL sample.
The elution buffer A (95/5, v/v, 10 mM ammonium acetate/acetonitrile, pH = 6.8) and the elution buffer B (20/80, v/v, 10 mM ammonium acetate/acetonitrile, pH = 6.8) were reasonable for effective chromatography and sensitive mass spectrometric detection. The separation elution gradient started at 100% eluent A for 5 min and increasing eluent B to 50% (v/v) within 55 min. Further on, in between 15 min the elution buffer B content was increased to 100% and held for 5 min. Finally, the mobile phase was reset to starting conditions within 5 min and the column was equilibrated before the next injection.
The HPLC solution from the initial 5 min of each run was collected into a separate vial (i.e., total volume of 100 μL) directly after UV/vis detection. After 5 min, the outlet capillary was connected to the ESI entrance and the eluting liquid was analysed by MS.
Several single and tandem MS experiments were performed in positive detection mode. All samples were primarily analysed, using single MS mode. In this mode all detectable substances could be received. MS/MS spectra were measured using the "survey scan" mode, switching from MS (m/z 50–1500) to MS/MS (m/z 50–1500) for intensive m/z values and backspaced to MS at a threshold of 100 counts.
The ion source temperature for all experiments was set to 80 °C and the desolvation temperature to 150 °C. The capillary voltage was 3.0 kV, and the cone voltage was 100 V. The collision energy ramp for MS/MS was set to 30/40/60 eV. Nitrogen was used as gas for drying and nebulization and argon as the collision gas for MS/MS.
Comparative study of literature using three databases: To determine the frequency of the terms "mass spectrometry", "HPLC", "molecular mass", "hydrophobicity" in relation to "proteomics" and "metabolomics", a literature search was performed for the presence of these combined terms in the title or abstract from 1 January 1997 to 10 September 2008. The databases used were ScienceDirect (SD),17 PubMed (PM)18 and Scopus (S)19 The amount of publications containing "proteomics" was 3216 (in SD), 8888 (in PM) and 26041 (in S) or for "metabolomics" 391 (in SD), 790 (in PM) and 1894 (in S). The three databases contain a different amount of journals which explains the different numbers.
The amount of relevant publications that listed "molecular mass" or "hydrophobicity" in relation to"proteomics" or "metabolomics" was also investigated. Finally, a publications quotient was calculated (as a percentage) for the frequency of "hydrophobicity" compared with "molecular weight" in relation to "proteomics" and "metabolomics". These values reflect the occurrence of keywords for the complementary techniques HPLC and MS in biological research.
Results and Discussion
This article is a review backed up with new data. The systematic research of very complex biological samples using LC–API-MS/MS will be discussed. It is, therefore, essential to initially look separately at the single features of the coupled techniques (HPLC, ionization and mass spectrometry). The information must then be put together to formalize an overall picture. This involves the parameter hydrophobicity, molecular weight and structure for the identification and characterization of known and unknown substances. The sample chosen (spontaneously obtained and filtered mouse urine) will highlight the advantages and limitations of this approach.
Determination of Hydrophobicity using LC
RP-LC separations depend on the interaction of the analytes with the hydrophobic reversed-phase materials and also the solubility of the analytes in the mobile phase. The term "hydrophobicity" was formed in the 1980s and is defined as the property of being water-repellent.20
Several elution factors were determined on chromatographic columns by applying the corresponding retention times of derivatized amino acids.21 Numerous scientific groups separated essential amino acids by RP-LC (predominantly on C18 modified silica) after their chemical modification. The elution times were typically normalized to the amino acid glycine (which was set to 1.0). There are currently seven approaches to achieve such separations, mainly resulting from a different pH of the mobile phases (pH 2,22,23 pH 3,24 pH 7,22,24,25 or pH gradients26).
The initial approach of our study was the analysis of biological real life samples (spontaneously obtained mouse urine) without further preparation. LC–API-MS analysis should be appropriate because peptides, proteins and metabolites can be detected, as well as a number of other small and large organic molecules. The chromatographic separation, originally developed by Cowan et al. at pH 7.5, was modified24 and established at a flow-rate of 20 μLmin–1.
Figure 1(a) shows the example of a mouse urine sample with its UV/vis chromatogram and corresponding mass spectrometric total ion chromatogram (TIC) [Figure 1(b)]. Hydrophobicity is explained using this sample chromatogram.


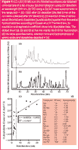
Figure 1
For the first five minutes the spectroscopic plotted chromatogram of the biological real sample [Figure 1(a)] shows a high signal, while these substances cannot be detected with mass spectrometry [Figure 1(b)]. This is due to the analytical assembly of the LC–MS instrumentation. The mobile phase is collected after the spectroscopic detection and is, therefore, not transferred into the mass spectrometer. This prevents mass spectrometer contamination by non-retained substances (such as salts) and at the same time the compounds can be collected and re-analysed later. In the first five minutes amino acids elute [see Figures 1(c) and (d)], as well as salts and polar urine components.
These amino acids elute after derivatization with an aromatic group in a second run (Scheme 1) within 38–60 min (Figure 2). This strategy of derivatizing polar molecules is a reasonable amendment to direct analysis using C18 modified LC materials. For the analysis of other substances, which are not retained in the column (for example, oligosaccharides) other derivatizing approaches were possible,27 but will not be discussed here.

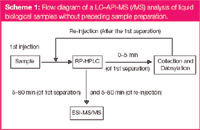
Scheme 1: Flow diagram of a LCâAPI-MS (/MS) analysis of liquid biological samples without preceding sample preparation.
The first observations of hydrophobicity were classically performed with modified amino acids.22–26 This classical approach to determine hydrophobicity is depicted with an example using dabsylated amino acids in Figure 2. Often, the derivatization can be achieved by a nucleophilic substitution, for example, at a sulphonyl–sulphur atom of dabsyl chloride. In this process the amino acid and peptides form sulphonic esters and sulphonamides with their alcohols and amines, respectively,28 holding a dabsyl moiety with aromatic configuration.16 Aromatic groups (such as dabsyl- and dansyl- etc.) were used as chromophores to enhance detection for UV/vis detectors.29 This is not necessary if mass spectrometric detection can be used. The chromaphore often represents a hydrophobic (mostly aromatic) skeletal structure, therefore, the well-established reaction methods16 can be used to increase the commonly low hydrophobicity of amino acids. The result is a significant extension of retention times for the initially non-retained amino acids [Figure 1(c)]. Figure 2 shows a plot of retention times of dabsylated amino acids versus hydrophobicities published by Cowan et al. (filled rhombus).24 The hydrophobicities of derivatized amino acids correlate very well with the published data (except arginine and lysine).This demonstrates that the original data, even after three decades, are useful for modern chromatography.

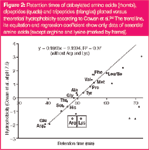
Figure 2
Substances from the original mouse urine that elute after five minutes were directly transferred into MS and analysed (Scheme 1). Figure 2 shows that analysed dipeptides with hydrophobicities according to Cowan et al., greater than 1.0, have elution times longer than five minutes and can be analysed without derivatization. At this point it should be stated, in contrast to these dipeptides, amino acids with hydrophobicities > 1.0 elute within five minutes. This shows that the hydrophobicity not only depends on the species of the amino acid but also on the chain length and particular effects, such as secondary structure.30 This effect was observed indirectly in this study. While retention times of dipeptides in Figure 1 (slightly time-shifted) fit well with the theoretical hydrophobicity (filled quads) some of the sterically demanding dipeptides differ a lot, and the tripeptides (filled triangles; Figure 2), as well as larger peptides, have no dependency at all (data not shown).
Generally, the retention time (i.e., hydrophobicity) of individual compounds is always determined in each chromatographic experiment. Hydrophobicity is seldom used to characterize unknown molecules, which is unfortunate because this parameter can be easily installed as a reference parameter for molecule specification.
During the last few years, some data sets have been published30–34 with biological molecules which can be coupled with mass spectrometric results from biological analysis. In this publication dozens of small peptides from "trypsin-digested" proteins were surveyed and their hydrophobicity was established by so-called retention coefficients.
Consequently, accurate predictions of retention behaviour of unknown peptides may be possible using these databases.
Unfortunately, these databases have to be re-established for each separation method in each separation lab. For the universal use of hydrophobicity all laboratories must use the same set of molecules for the creation of standard databases at all times. Tryptic digests of frequently applied proteins (e.g., bovine serum albumin or lysozyme) can be used. The easiest way forward would be to adapt a published separation and use it in all laboratories. This would not be widely accepted because of the legitimate conflict of interests between different column manufacturers, so the use of digests is a more realistic alternative.
This approach should be appointed by national and international directives and become obligatory for publications. In line with "Good Laboratory Practice" (GLP),35 this set of hydrophobicity standards can, for example, be integrated into open-source software frameworks such as "OpenMS".36 This standardization would allow the use of the parameter hydrophobicity to validate collected MS data (and vice versa).
Furthermore, the hydrophobicity parameter obtained should be transferred to analytes of other chemical groups, whose general structure is not based on amino acids. The use of hydrolysed peptides is recommended for the future as the analysis is simpler.30–34 This would help to assign other molecules because the peptides can cover a complete chromatographic run. Here, once again, we call on competent authorities (such as DIN or EU) to create a standardized set of molecules.
Determination of Molecular Weight Using MS
As well as retention time, the complementary parameter of molecular weight can be obtained for various analytes using LC–MS approaches. The mobile phase of the chromatographic separation was directly transferred into the mass spectrometer — in the current example from the fifth minute up to the end of that run — (Scheme 1). The applied urine sample contained analytes with low hydrophobicity eluting in the first five minutes and moderately to strongly hydrophobic analytes with elution times longer than five minutes. The total ion chromatogram (TIC) in Figure 1(b) shows that a lot of contained substances can be separated with C18-modified silica material (with more or less sufficient chromatographic resolution).
This separation efficiency would not be high enough for separations with UV/vis detection [e.g., Figure 1(a)], as co-eluting components overlay with their particular absorption. However, using mass spectrometric detection does not necessarily require a chromatographic baseline resolution [Figure 1(b)], because the compounds could be detected as single ions via their specific mass-to-charge ratio, which are often independent from other co-eluting components. In chromatography this leads to moderate run times but also inevitably to smaller specificity of their hydrophobicities. Nevertheless, hydrophobicity ensures — as pointed out in the last section— the identification of substances from unreliable mass spectrometric information. Such unreliable information can be expected, if one wants to identify substances using a working mass spectrometer with low accuracy (such as a quadrupole or an ion trap instrument).
Types of Mass Spectrometer
This section will describe the types of mass spectrometer currently available and their application in bioanalysis with an emphasis on mass spectrometric parameters such as resolution, sensitivity and accuracy.
Single quadrupole mass spectrometer (Q): These relatively inexpensive instruments can mainly be used for the detection and quantification of known molecules. This equipment should be used for characterizing molecules, when their reference materials are available. Quadrupole units only cover a small mass range at high sensitivity with relatively low resolution and accuracy.
However, one can analyse many ionized substances sensitively using LC–Q-MS with recent time-set units at a narrow mass range for each molecule. Furthermore, because of fast polarity-switching mode, substances can be detected very sensitively, even at different polarities of the detectable ions. The fast polarity change of Q-units, therefore, enables a quasi-simultaneous detection of positive and negative charged molecules. This detection feature had been limited to Q-units, but it is also used in a time-of-flight (ToF) mass spectrometer. Some of the advantages of determining ions of different polarity are listed in the section titled "Ion Sources and Polarity Switch for Specific Ionization".
Single time-of-flight mass spectrometer (ToF): These instruments are used to characterize known molecules, but more often to determine molecular weight of unknown molecules. Recent units allow the molecular weight of very large and highly loaded molecules to be determined because of their high mass spectrometric resolution and the excellent mass accuracy. A mass spectrometric resolution of 10000–19000 was, until now, the maximum for routine acquisition of large molecules as proteins. Meanwhile, high-resolution ion trap instruments such as Orbitrap and FTICR have beaten this record with possible resolutions above 100000.
With a ToF instrument many ions can be measured simultaneously without losing sensitivity. Faster data handling of detector signals using modern computers and software will propel this advantage.
A TIC originated from a ToF as shown in Figure 1(b) for the m/z range of 50–1500 can be presented in a more detailed way and can be evaluated with a small amount of effort. Figure 3 shows exemplarily extracted ion chromatograms (EICs) for five mass spectrometric traces. The more accurate the molecular masses that are determined, the narrower the particular mass spectrometric range that can be chosen and the more precise mass traces that can be assigned to molecules. Thus ToF-MS permits a mass accuracy down to 0.00(0)1 Da, whereas newer types Q-MS enable accuracies to 0.1 Da. Therefore, in this instance only ToF instruments are suitable for determining empirical formula for biomolecules up to a molecular weight of 1000 Da.

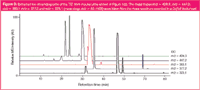
Figure 3
It is often a question for the depiction of EICs, whether simultaneously detected mass traces result from only one molecule or from co-eluting components. The coincidence of the molecular traces can easily be differentiated. In Figure 4, the assay of the top EIC from Figure 3 (m/z = 428.3) with their two signals at 22.1 min and 24.1 min [Figure 4(a)] is specified in detail. Figure 4(b) shows the corresponding base mass spectra of mouse urine at a maximum signal of EICs m/z = 428.3. The left spectrum has three mass signals (m/z = 226.2, m/z = 428.3 and m/z = 444.3), a fourth m/z = 257.2 adds to the same three signals in the right spectrum. Again, the single mass traces of the spectrum are shown as EIC and the particular curves are compared by overlay [Figure 4(c)]. The Figure clearly addresses the question of ion origins with a single mass spectrometer. For this example it is quite clear that in both chromatographic signals the molecular masses m/z = 226.2, m/z = 428.3 and m/z = 444.3 belong to one molecule. If the mass traces emanate from different molecules, the curve shape (as m/z = 257.2) would be different. Although both signals obviously represent the same molecule, retention times differ significantly. This may be a result of the presence of an isomeric structure, where the later-eluting molecule has a higher hydrophobicity compared with the other molecule (see also section titled "Ion Sources and Polarity Switches for Specific Ionization")

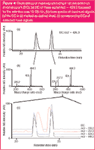
Figure 4
Once again, the difference between Q- and ToF-instruments is clearly depicted by the ion masses m/z = 428.3 and 444.3. The difference of both [in Figure 4(b)] lettered molecular masses is 16.0 Da; this could correspond to a loss of oxygen or methane (CH4). A Q-MS unit could not accomplish the assignment, whereas using a mass calibrated ToF-MS unit allows the clear assignment of oxygen as the lost atom.
Other mass spectrometers: Molecular mass determination of large and smaller molecules can also be achieved using other mass spectrometers. Ion trap instruments can achieve both determination of molecular mass and structure. Fourier transform ion cyclotron resonance mass spectrometers (FTICRs) and Orbitrap mass spectrometers reflect the latest development in mass spectrometric detection. With their extremely high resolution and good accuracy analysts can probably achieve the best results in evaluation of molecular masses by far. Also, the previously mentioned loss of 16 Da would have been unambiguously assignable with these instruments. Unfortunately, only special laboratories possess such equipment because of the high price.
Other ion trap mass spectrometers lie somewhere between quadrupole and ToF-units. Accordingly, they are more commonly used to characterize (e.g., quantify) known molecules. The strong point of ion trap instruments is their ability to identify unknown molecules using MSn mass spectrometry. These instruments are also suitable for qualitative and quantitative analysis.
Determining Molecular Structure Using Tandem Mass Spectrometry
The molecular structure can be observed by using LC-tandem MS, as well as the determinination of elution characteristics and the molecular weight of single components. Figure 5 explains the appropriate procedure. An EIC from Figure 3 was chosen again. The broad, asymmetrical signal m/z = 447.2 [Figure 5(a)], and the corresponding mass spectrum is depicted at the signal maximum [Figure 5(b)]. The extracted ion chromatograms [Figure 5(c)] indicate that both signals m/z = 447.2 and m/z = 271.1 cohere, whereas the mass signals m/z = 301.3 and m/z = 346.3 indicate two independent molecules. In this instance both could be verified by MS/MS measurements.


Figure 5
Collision induced dissociation (CID): In tandem MS (MS/MS) in the "first mass spectrometer" an ion with specific m/z is selected (e.g., m/z = 447.2), which is fragmented in a collision cell filled with inert gas [for collision induced dissociation (CID)]. Subsequently, the fragments are separated and detected in a "second mass spectrometer" [Figure 5(d)]. This fragmentation method has been mostly used in MS/MS and has been integrated into MS/MS instruments such as:
Ion trap mass spectrometer (IT-MS): IT instruments are offered with different geometrical design and can be coupled to other mass analysers. These units, using MSn-mode are called "over time" tandem mass spectrometers, because fragmentation happens in chronological order. Ions are first collected in an MS cell and fragmented by gas collision. This fragmentation is followed by accumulation of one of the resulting fragment ions, which again can pass through the same process. These instruments are especially suited to fragmenting complex molecules with many functional groups and to downscale their structure.
Triple quadrupole mass spectrometer (QqQ): These instruments are a sort of classical tandem mass spectrometer. For normal analysis of molecules they have the same features as single quadrupole instruments and are, therefore, only of limited use for unknown biomolecules. Their potency is the high sensitivity towards determination of molecule fragments for quantification of low concentrated substances from complex biological matrices. There is also a new combination of QqQ-linear IT-MS, combining all advantages of single analysers. For a detailed review on using QqQ-linear IT-MS see the recommended recently published book.37
Quadrupole time-of-flight mass spectrometer (QqToF): For bioanalysis these instruments are currently the most well-established systems for analysing large and small molecules. Here, as with QqQ instruments, "Q" stands for a real quadrupole and "q" for quadrupole as the collision cell for CID. These mass spectrometers combine the advantages of a continuous quadrupole with the resolution and accuracy of a ToF analyser. This means, these systems can analyse molecules and their fragmentation very accurately. Figure 5(d) shows some spectra of tandem MS measurements using such a QqToF instrument. Here m/z = 447.2 has been extracted and fragmented. Additionally, m/z = 271.1 a m/z = 243.0, 215.0 and 197.0 were generated. The mass differences are 176.1 Da, 28 Da and 18 Da, whereas single fragments coercively come from m/z = 271.1 and not from other fragments. For the latter conclusion the MSn analysis using IT would be required. During our experiments many substances were fragmented and some could finally be identified using QqToF. Within the scope of this article individual results will not be described further. It should be mentioned, however, that although many signals derive from peptides and proteins, just as many derive from different organic classes, such as sugars and metabolites.
Molecular fragmentation techniques: Normally fragmentation is accomplished in collision cells (as mentioned previously, commonly a quadrupole) to achieve information on structure, but a lot of other fragmentation technologies were launched recently, some of which differ extremely in fragmentation specifities compared with CID. That is why CID now competes directly against those techniques: "blackbody infrared radiative dissociation" (BIRD), "electron-capture dissociation" (ECD), "electron transfer dissociation" (ETD) and "infrared multiphoton dissociation" (IRMPD).
Principally, one can of course induce fragmentation of molecules with single quadrupole and ToF instruments. Then it can benefit from the so-called fragmentation within the "ion source", that is, at the entrance area of the mass spectrometer (cone-voltage driven and fragmentor-voltage driven). This seems reasonable, especially if the samples include single components, or substances that elute with baseline resolution from the column. Unfortunately, this is normally of little importance, as non co-elution with other molecules from biological samples seldom happens. For a more profound and detailed treatment of mass spectrometry with all its facts, we recommend three recently published books.37–39
Ion Sources and Polarity Switch for Specific Ionization
All results achieved so far were performed using the ionization technique, electrospray ionization (ESI), in positive detection mode. Consequently, this ionization method is more an ion transfer from LC's mobile phase to the gas phase and, accordingly, postulates that molecules already exist charged in solution. For uncharged and chemically stable molecules, real ionization techniques, such as the atmospheric pressure chemical ionization (APCI)40,41 and the atmospheric pressure photo ionization (APPI)41,42 can be applied. This ionization is especially useful for small molecules such as metabolites from biological samples. "Labile" molecules, such as proteins, are simultaneously disrupted, thus a different selectivity can be achieved with different ion sources. Finally, APCI and APPI molecules can be detected, which would have been lost using LC–ESI-MS. For example, organic molecules used as pharmaceutical substances,42 organic dyestuffs43 and partially oxidized polycyclic aromatic hydrocarbons (PAHs).40,41,44
The latter publications showed a clear dependency of retention time and mass-to-charge ratio of detected ions to their polarity and included functional groups. In these studies higher hydrophobicity was obtained for aromatic compounds containing two adjacent hydroxyl groups. Not only was the hydrophobicity influenced by the resulting hydrogen bond (and therefore the corresponding pseudo aromatic status), but electrons were also preferentially captured from these molecules. Hence, the negative MS mode received these molecules mainly as negative molecular ions [M]– and not as the general deprotonated molecular ions [M–H]–.44
Several molecules can be — because of their chemical properties — simultaneously detected as negative and positive ions. This information should also be used for a better characterization of unknown molecules.44 This behaviour is particularly interesting for metabolite research. Metabolomics currently tends not to consequently identify each single component, but puts all available characteristics of compounds into databases for a final sorting. Thus the more molecular "parameter" that can be observed for single molecules, the easier the classification is.
Conclusions
For future investigations in LC–API-MS/MS we want to emphasize the importance of the chromatographic parameter, as well as mass spectrometric information. The ionization parameter can be established if APCI- and APPI-measurements become routine in biological samples and if the mass spectrometers can simultaneously detect positive- and negatively-charged substances. However, in reality the latter two detection properties will not be routinely used because of the limited amount of mass spectrometers with these options.
By considering the three parameters — hydrophobicity, molecular weight and molecular structure — as well as ionization properties, each detected molecule will become a unique molecule. Furthermore, these "uniques" can be stored in databases of corresponding analysis systems and be used for cross-laboratory identification and characterization of these molecules. Such databases would be linked by reference molecule sets and could be used universally.
By reviewing the amount of scientific publications in bioanalysis for "proteomics" and "metabolomics" published till now, one can see that the hydrophobicity can become very important.. The term "hydrophobicity" occurs one tenth less than the term "molecular mass" in proteomics' publications (exactly 6% in SD, 7% in PM, 9% in S. The amount of times it appears in metabolomics' publications is 0% in SD, 5% in PM and 5% in S.
The ionization parameter can be established, if APCI- and APPI-measurements become routine in biological samples, and if all mass spectrometers are able to detect positive and negatively charged substances simultaneously. However, this parameter is only of limited value, because both topics will not become routine soon.
Acknowledgment
Our thanks go to the German Research Foundation and the Chair of Biopolymer Chemistry for financial support. We thank I. Frey and Prof. H. Daniel for numerous reference materials as well as mouse urine, Prof. H. Klostermeyer and Prof. L. Moroder for many peptide standards and I. Krause for detailed information on dabsylation.
Christiane Berkemeyer worked as a chemical laboratory assistant in the "Analytical Research Group" at the Chair of Biopolymer Chemistry, TU München. Meanwhile, she has been working on solving analytical problems in the Swiss industry.
Thomas Letzel is assistant professor at the TU München in Freising/Bavaria/Germany and head of the "Analytical Research Group" at the Chair of Biopolymer Chemistry. His research interests are currently focused on fundamental mass spectrometric studies and developing new tools in "functional proteomics".
References
1. J.C. Smith et al., Anal. Chem., 79, 4325–4344 (2007).
2. M. Kussmann et al., Mass Spectrom. Rev., 26, 727–750 (2007).
3. L. Alder et al., Mass Spectrom. Rev., 25, 838–865 (2006).
4. T.A. Brettell, J.M. Butler and J.R. Almirall, Anal. Chem., 79, 4365–4384 (2007).
5. Y. Zhang, G. Zhang and Y. Zangh, J. Chromatogr. A, 1075, 1–21 (2005).
6. A. Drabik et al., Mass Spectrom. Rev., 26, 432–450 (2007).
7. R.K. Gilpin and C.S. Gilpin, Anal. Chem., 79, 4275–4294 (2007).
8. C. Zwiener and F.H. Frimmel, Anal. Bioanal. Chem., 378, 862–874 (2004).
9. S.D. Richardson, Anal. Chem., 79, 4295–4324 (2007).
10. G.A.D. Souza, L.M. Godoy and M. Mann, Genome Biol., 7, R72 (2006).
11. J. Adachi et al., Genome Biol., 7, R80 (2006).
12. J.H. Weiner and L. Li, BBA-Biomembranes, 1778, 1698–1713 (2008).
13. B. Pilch and M. Mann, Genome Biol., 7, R40 (2006).
14. EN ISO 9001:2000; ISO/IEC 17025.
15. European Commission; Implementing Council Directive 96/23/EC concerning the performance of analytical methods and the interpretation of results. Official J. L. 221, p. 8–36 (2002).
16. I. Krause et al., J. Chromatogr. A, 715, 67–79 (1995).
17. http://www.sciencedirect.com, (September 10, 2008).
18. http://www.ncbi.nlm.nih.gov/sites/entrez, (September 10, 2008).
19. http://www.scopus.com, (September 10, 2008).
20. http://www.thefreedictionary.com/hydrophobicity, (September 10, 2008).
21. http://www.expasy.ch/tools/protscale.html, (September 10, 2008).
22. J.L. Meek, Proc. Natl. Acad. Sci. USA, 77, 1632–1636 (1980).
23. C.A. Browne, H.P.J. Bennett and S. Solomon, Anal. Biochem., 124, 201–208 (1982).
24. R. Cowan and R.G. Whittaker, Peptide Res., 3, 75–80 (1990).
25. J.M.R. Parker, D. Guo and R.S. Hodges, Biochemistry, 25, 5425–5431 (1986).
26. K.J. Wilson et al., Biochem J., 199, 31–41 (1981).
27. D. Cointe, Y. Leroy and F. Chirat, Carbohydr. Res., 311, 51–59 (1998).
28. J. March, Advanced Organic Chemistry, 3rd ed., John Wiley & Sons, New Jersey, USA (1985).
29. J.-K. Lin and C.-C. Lai, J. Chromatogr. B, 227, 369–377 (1982).
30. Y. Wang et al., J. Chromatogr. B, 826, 122–128 (2005).
31. M. Palmblad et al., Anal. Chem., 74, 5826–5830 (2002).
32. M. Palmblad et al., J. Chromatogr. B, 803, 131–135 (2004).
33. M. Schweizer et al., Food Chem., 105, 1606–1613 (2007).
34. B. Tripet et al., J. Chromatogr. A, 1141, 212–225 (2007).
35. R.D. McDowall, LCGC Eur., 21(6), 1–10 (2008).
36. M. Sturm et al., BMC Bioinformatics, 9, 163–173 (2008).
37. K. Wanner and G. Hoeffner, Mass Spectrometry in Medical Chemistry, Wiley-VCH, Weinheim, Germany, (2007).
38. I.A. Kaltashov and S.J. Eyles, Mass Spectrometry in Biophysics, John Wiley & Sons, New Jersey, USA, (2005).
39. H. Rehm, Der Experimentator: Proteinchemie/Proteomics, 5th ed., Elsevier GmbH, Munich, Germany, (2006).
40. T. Letzel et al., Anal. Chem., 73, 1634–1645 (2001).
41. C. Adelhelm et al., Anal. Bioanal. Chem., 391, 2599–2608 (2008).
42. A. Raffaelli and A. Saba, Mass Spectrom Rev., 22, 318–31 (2003).
43. E. Rosenberg, Anal. Bioanal. Chem., 391, 33–57 (2008).
44. S. Grosse and T. Letzel, J. Chromatogr. A, 1139, 75–83 (2007).








Study Explores Thin-Film Extraction of Biogenic Amines via HPLC-MS/MS
March 27th 2025Scientists from Tabriz University and the University of Tabriz explored cellulose acetate-UiO-66-COOH as an affordable coating sorbent for thin film extraction of biogenic amines from cheese and alcohol-free beverages using HPLC-MS/MS.

Quantifying Microplastics in Meconium Samples Using Pyrolysis–GC-MS
March 26th 2025Using pyrolysis-gas chromatography and mass spectrometry, scientists from Fudan University and the Putuo District Center for Disease Control and Prevention detected and quantified microplastics in newborn stool samples.

