Retention Time Prediction Based on Molecular Structure in Pharmaceutical Method Development: A Perspective
LCGC North America
The principal aim of this work was to provide a perspective with practical utility in streamlining the chromatographic method development in pharmaceutical industries based upon predicting the chromatographic retention times from molecular structures. Workflows were suggested with a focus on reversed-phase LC, IC, and HILIC as the three major techniques. Unlike HILIC, retention prediction in both reversed-phase LC and IC can benefit from the maturity of these techniques and the transparency of their retention mechanisms. In reversed-phase LC the solute coefficients in the hydrophobic subtraction model and in IC the a and b values in the linear solvent strength model can be the subject of modelling with their subsequent use in retention prediction. A workflow for HILIC can be based on the design of experiments approach, to account for all major contributors to the retention mechanism, and direct correlation of experimental retention times to the molecular descriptors.
The principal aim of this work was to provide a perspective with practical utility in streamlining the chromatographic method development in pharmaceutical industries based upon predicting the chromatographic retention times from molecular structures. Workflows were suggested with a focus on reversed-phase liquid chromatography (LC), ion chromatography (IC), and hydrophilic-interaction chromatography (HILIC) as the three major techniques. Unlike HILIC, retention prediction in both reversed-phase LC and IC can benefit from the maturity of these techniques and the transparency of their retention mechanisms. In reversed-phase LC the solute coefficients in the hydrophobic subtraction model and in IC the a and b values in the linear solvent strength model can be the subject of modeling with their subsequent use in retention prediction. A workflow for HILIC can be based on the design of experiments approach, to account for all major contributors to the retention mechanism, and direct correlation of experimental retention times to the molecular descriptors.
Pharmaceutical analysis using multiple chromatographic methods is required during most stages of pharmaceutical method development. There are a number of chromatographic techniques that are readily amenable for small-molecule pharmaceutical analysis, including gas chromatography (GC), liquid chromatography (LC), supercritical fluid chromatography (SFC), and electrokinetic separations. Figure 1 illustrates a conceptual “chromatographic space” that encompasses all pharmaceutically relevant chromatographic techniques and illustrates the extent of distinction and overlap among these techniques. The choice of a suitable chromatographic method of analysis generally proceeds through three stages. The first stage is to select the appropriate technique that is expected to provide the desired separation selectivity with adequate retention for the analytes of interest. This selection involves a choice not only of the major class of chromatography (for example, GC or LC) but also the specific chromatographic technique within that class. For example, while within the broad classification of LC reversed-phase high performance liquid chromatography (HPLC) is the most widely used purification and separation technique, other techniques including ion chromatography (IC) and hydrophilic-interaction chromatography (HILIC) are also used as complementary methods that offer different selectivities and retentions. The second stage is column screening or column scouting, which is usually an experimental phase aiming to identify the column (stationary phase) offering adequate resolution of analytes of interest. The third, and final, stage of method development is to identify the precise details of the separation conditions by inclusion and optimization of other instrumental parameters believed to affect the separation, such as the exact composition of the mobile phase, the flow rate, the temperature, and so forth.


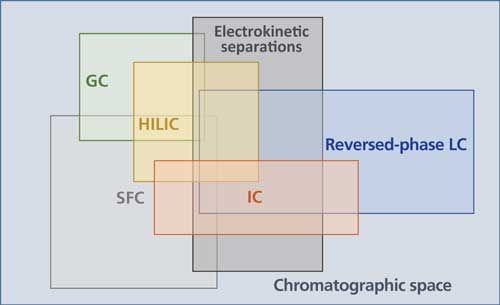
Figure 1: A definition of a conceptual chromatographic space.
At present, most of the method development in the pharmaceutical and other industries is carried out by trial-and-error laboratory experimentation that requires screening of multiple types of LC techniques and numerous chromatographic columns using a large number of experimental conditions. This trial-and-error approach is inevitably time-consuming and costly in generating a significant amount of waste and exhausting both human and instrumental resources. On the other hand, new advances in combinatorial chemistry have enabled pharmaceutical industries to synthesize a much greater number of potential new drugs than at any time in the past. As a result, analytical method development is also required to be fast enough to keep up with the high throughput of drug discovery processes. Additionally, developed analytical methods nowadays need to fall within a quality-by-design (QbD) framework. When applied to chromatographic method development, QbD principles dictate that the specific method developed for a particular separation should provide a continuous stream of appropriate data throughout the method lifecycle by maintaining optimal performance, even when there are slight changes in the conditions (1).
The emergence of new instrumentation, such as ultrahigh-pressure liquid chromatography (UHPLC) systems, together with advances in column technology have greatly improved efficiencies in method development. In addition, column characterization and classification methods are well-studied and used routinely to guide analysts to choose either similar or dissimilar stationary phases, depending on development needs. However, with ever more diverse stationary phases available, it also becomes more challenging for analysts to select a suitable chromatographic system or even a starting point for method development. Hence, method development is widely recognized to be the most time-consuming and costly phase of pharmaceutical analysis, and it has become a major goal for the industry to streamline this process.
Overview of Quantitative Structure–Retention Relationships
In an effort to minimize the number of experiments needed for analytical method development, one attractive solution is using the molecular structures of analytes for the rapid prediction of retention times at any given chromatographic conditions. Accordingly, the structural features of a given compound are used to derive physicochemical properties or some other numerical representation of the compound, which are generally called molecular descriptors. These molecular descriptors are then correlated with retention data through statistical modeling, and the entire process is referred to as quantitative structure–retention relationships (QSRR). Molecular descriptors can be computationally derived or determined empirically. Unlike computationally derived descriptors, the determination of empirical descriptors (also called solvatochromic parameters), which are used in approaches based on the linear solvation energy relationships (LSER), is elaborative and requires time-consuming experimentation. This limitation somewhat hinders the purpose of retention time prediction for rapid method development using the LSER approach.
Ideally, by comparison of the predicted retention results across available chromatographic techniques, one would be able to select the best starting point for method development. This primary phase of method development is called scoping and would involve selection of the preferred chromatographic technique, the preferred column (or column class), and the broad composition of the mobile phase (especially the preferred type of organic modifier, or ion-exchange competing ion, and so forth). Further optimization of the selected conditions can then be performed through a subsequent phase called optimization by implementing some form of experimental design approach, such as factorial designs or response surface methodologies, that can be found in statistical software packages such as Modde (Umetrics, Inc.), Design-Expert (Stat-Ease, Inc.), JMP (SAS Institute, Inc.), and Fusion Pro (S-Matrix, Corp.). A typical workflow summarizing these two phases is illustrated in Figure 2.


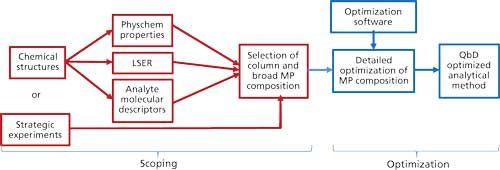
Figure 2: A workflow of systematic chromatographic method development.
Since their introduction in the late 1970s (2–4), QSRRs have gained a great deal of interest and have been studied for chromatographic retention predictions. As can be inferred from its name, in QSRR retention is modeled as a function of molecular descriptors, which are the numeric transformations of molecular structures. As mentioned earlier, the descriptors can be either experimentally determined or theoretically computed. Indeed, commercial software packages such as Dragon (Talete, srl) enable computation of thousands of descriptors for virtually any chemical entity. Typically in a QSRR study, retention times for some representative compounds (that is, a training set) are determined experimentally under a specified chromatographic condition and used to correlate with the most informative molecular descriptors. In this context, retention time (response) serves as a dependent variable, while molecular descriptors serve as independent variables. After a statistically significant mathematical model relating retention time to relevant descriptors is established, retention times of new compounds can be predicted for the given chromatographic system, based only on the molecular descriptors included in the model and without the need for additional experimentation.
To contribute to streamlining the analytical method development process, a QSRR study should be ideally in the form of a software platform that can be used for the following steps:
- identify the most suitable chromatographic method for the set of analytes in hand,
- enable prediction of retention time with enough accuracy for several chromatographic conditions,
- enable optimization of the selected chromatographic conditions with the minimum of extra experimentation, and
- ensure the highest possible quality of the final method in terms of robustness, resolution, run time, and so on.
Despite numerous studies carried out in QSRR modeling, such a comprehensive QSRR study with an immediate practicality in pharmaceutical method development is still lacking. In one study QSRR models were built for a dataset containing 62 diverse compounds analyzed under 75 chromatographic conditions (15 columns and five reversed-phase LC gradient elutions with different slopes). However, except for the list of the top 20 most relevant descriptors, no other important information was declared, including the chemical structures of 48 compounds, presumably because of proprietary reasons (5).
In the following discussion, we present a brief perspective for a more effective implementation of QSRR in pharmaceutical method development, with a focus on reversed-phase LC, IC, and HILIC as three of the most widely used LC techniques in the pharmaceutical industry.
Method Development in Reversed-Phase LC
Reversed-phase LC is by far the most prevalent liquid chromatography technique for pharmaceutical analysis, where the high dynamic range and low variability of instruments, together with the compatibility of most formulations and active pharmaceuticals with its separation condition, allow for a majority of assays to be performed. Not surprisingly, a great deal of effort has also been devoted to predicting chromatographic retention times under reversed-phase conditions.
There are several approaches to the prediction of retention times based on chemical structures, and some of these have even been embedded into commercial software packages. For example, one commercial tool simply uses molecular molar volume and energy of interaction with water as two preselected descriptors to build QSRR models (6). Another simple QSRR model proposed by Baczek and Kaliszan correlates reversed-phase LC retention time to the total dipole moment, the electron excess charge of the most negatively charged atom, and the water-accessible molecular surface area of the analyte (7). Both of these models, together with some other simple models proposed earlier, take a simplistic view of the reversed-phase LC retention mechanism, particularly by neglecting the ionization state of the compounds, and therefore provide only a moderate prediction accuracy under a given set of conditions. Relative prediction errors in the range 19–27% in retention factor, k, were obtained with the latter model (7). The target prediction error for a model having practical utility in retention prediction is expected to be less than 5% in k (7).
There is another commercial tool designed for retention time prediction based on physicochemical properties of target analytes that have been calculated for a set of “training” compounds in advance, and are stored in a knowledge base. These physicochemical parameters include logP, logD, polar surface area, molecular volume, molecular weight, molar refractivity, and the number of hydrogen bond donor and acceptor sites on the molecule. A multiple linear regression (MLR) model is then built to correlate these parameters to the experimental retention data. For any new analyte the above mentioned parameters are calculated and then are fitted to the model equation to generate the predicted retention time. One unique feature of this tool is the use of a rather different strategy for modeling named federation of local models. In this approach instead of using a “global” model built using all the compounds in the knowledge base for predicting the retention time of any given compound, a locally predictive model is established using only those compounds in the knowledge base that are most structurally similar to the compound of interest (6). Consequently, the prediction accuracy for any given compound would depend strongly on the number of similar compounds (usually 20) that can be found in the knowledge base, as well as their level of similarity. Limitations in this tool lie in considering only a very limited number of physicochemical descriptors in modeling, which is necessarily a simplification of the retention mechanism that can affect the predictive quality of the models, and also in overlooking the change in both the pKa of analytes and the effective pH of the buffer because of the presence of organic modifiers (6).
As an alternative to the use of a limited number of physicochemical descriptors, one can generate a very large pool of molecular descriptors (which include the physicochemical properties) using a molecular modeling software such as Dragon. Because of the large number of descriptors generated (typically more than 4000), it is then necessary to extract the most informative descriptors by implementing a suitable variable selection method, such as a genetic algorithm (GA), combined with a multiple regression method, such as MLR or partial least squares (PLS), to generate the model. Although it is apparent that the most informative descriptors are those resulting in the most statistically significant model for prediction, one may wonder what would be the optimum number of descriptors in a model. As mentioned earlier, a model with too few descriptors could be not predictive enough (under-fitted), whereas too many descriptors can bring noise to the model and increase the risk of over-fitting and chance correlation (8). Unlike MLR, PLS is known for being particularly useful in handling datasets with a much greater number of variables than samples and in the presence of collinear, redundant, or noisy variables. However, studies have also shown that even the performance of a PLS model could be substantially improved by incorporating a suitable variable selection method into the modeling (9,10).
Figure 3 illustrates the correlations obtained between experimental and predicted retention times for 14 compounds selected from a dataset containing 86 suspected sports doping species and nearly 1000 molecular descriptors generated by Dragon (10,11). Retention time prediction was performed using PLS models derived from all descriptors (Figure 3a) or from the most informative descriptors selected by implementing a GA-PLS algorithm (Figures 3b and 3c). While the goodness of fit and the mean absolute error (MAE) obtained with the two approaches appeared to be comparable, GA-PLS was found to be quite effective in significantly reducing the number of descriptors used for modeling by more than 90%. To explore the effectiveness of the federation of local models approach in enhancing the prediction accuracy, locally predictive PLS models were also established using subsets of only the 20 most structurally similar compounds to each compound and used for their retention time prediction. As can be seen in Figure 3c, great improvements in both the goodness of fit and the MAE were achieved, suggesting the critical role in prediction accuracy of the similarity between the dataset used for modeling and the compound of query. Apparently, the predictive quality of any QSRR model depends on how comprehensive the selected descriptors are in projecting the retention mechanism involved. In fact, retention prediction in reversed-phase LC can greatly benefit from the maturity of this technique and the transparency of its retention mechanism.


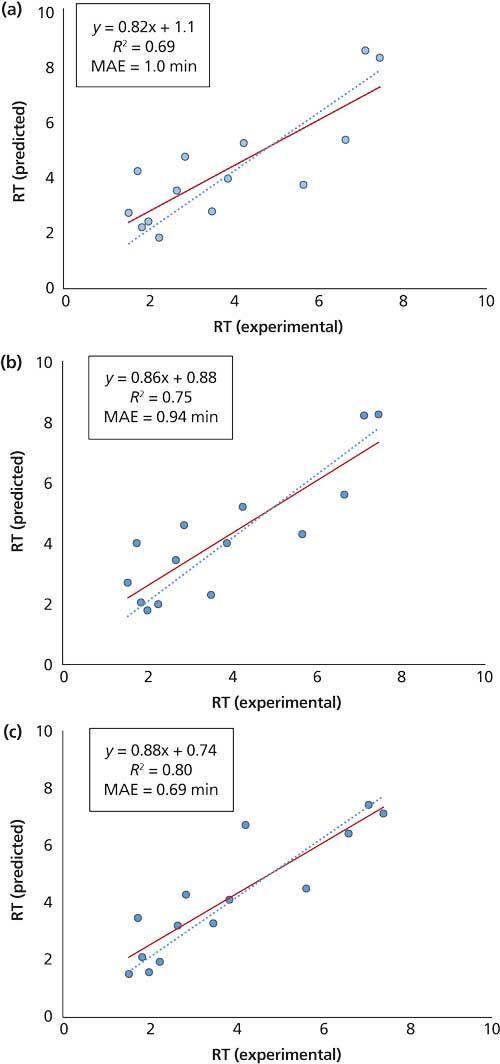
Figure 3: Correlation obtained between experimental and predicted retention times of reversed-phase LC data using (a) a PLS model with all descriptors, (b) a PLS model derived from descriptors selected by GA-PLS, and (c) an individual PLS model for each compound using descriptors selected by GA-PLS and the top 20 most similar compounds.
Being based on a LSER approach, the hydrophobic-subtraction model proposed by Snyder and colleagues (12) has been proven to account for all major contributors in the reversed-phase LC retention mechanism:


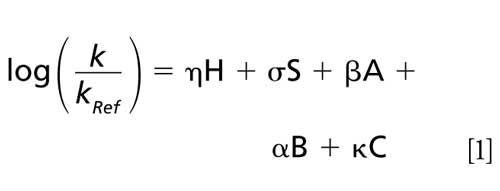
In this equation, the retention factor k of a given solute (relative to the reference solute ethylbenzene) is correlated with the complimentary properties of the solute and the column. The solute coefficients are denoted by small Greek letters, where η refers to solute hydrophobicity, σ to solute bulkiness, β to solute hydrogen-bond basicity, α to solute hydrogen-bond acidity, and κ is the effective charge on the solute molecule. The corresponding column properties are denoted by capital letters and determine the selectivity of the most commonly used reversed-phase LC columns (13). It is worth noting that the first four column properties are relatively independent of the mobile phase conditions, whereas the C-term is pH dependent and available at two pH values (2.8 and 7.0) (14).
Although equation 1 was proposed primarily for the purpose of classification of reversed-phase LC stationary phases based on differences in their selectivity, a remarkable prediction accuracy of ±1% in k was obtained for 150 different solutes and 90 type-B alkylsilica columns (13), suggesting its suitability for the purpose of retention time prediction as well. In fact, one of the unique advantages of using this equation for retention prediction is that a large fraction of all commercial reversed phases have been studied before and the column coefficients for almost every reversed-phase LC stationary phase from all major manufacturers have been collected in an on-line database hosted on the United States Pharmacopeia (USP) website (15). To date, this database contains column coefficients for more than 670 columns and is updated regularly.
Unfortunately, despite its prediction accuracy, the usefulness of this model for routine usage is limited, chiefly because the determination of the required solute coefficients for a new solute would require extensive experimental effort, which somewhat defeats the purpose of retention time prediction (12).
As an alternative to experimental determination of solute coefficients for every given solute, one approach could be their prediction from a set of individual models, any one of which correlates a solute coefficient as the dependent variable with some molecular descriptors. Put simply, the retention time for a given compound would be calculated from its predicted solute coefficients paired with their already existing column coefficients via equation 1. This approach requires a careful selection of some representative compounds as the training set, for which the solute coefficients must be inevitably determined experimentally. Once the representative models are established, the corresponding coefficients for any given compound could be determined without the need for any extra experimentation. If time and resources permit for expanding the training set, this approach could also benefit from the federation of local models strategy for improving the prediction accuracies even further.
Method Development in IC
In pharmaceutical drug development an appropriate and stable salt form for some drug substances is required for purposes such as improving the stability and water solubility of the drug and to assist in drug bioavailability. Inorganic ions, as well as small organic acids or bases, are often used as counterions of ionizable drug substances (16). Although IC is the primary method of choice for the determination of inorganic anions, it also has a great potential in analyzing other small ionic species by offering complementary selectivity to reversed-phase LC.
The retention factor, k, of a given ionic solute in IC under isocratic conditions can be simply correlated to the eluent concentration [E] using the linear solvent strength (LSS) model (17):


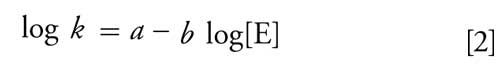
However, experimental measurement of a and b values is difficult since they are analyte-specific and also depend on some other parameters, such as the ion-exchange selectivity coefficient between the solute and the eluent competing ion, the ion-exchange capacity of the stationary phase, the weight of the stationary phase, and the volume of eluent in the column (18). Practically, a and b values for a given solute and a specific phase can be estimated on the basis of some experiments in which the retention factor is measured at a limited number of isocratic eluent concentrations and the resultant data are fitted to equation 2. This approach is the basis for retention time prediction and separation optimization in commercial software, which to date includes data for about 150 inorganic anions and cations, and carbohydrate species for 20 IC phases (19,20).
Although this tool is quite successful for rapid IC method development and optimization, it is designed to provide solutions for only the compounds that have been already included in its knowledge base, and therefore is not suitable for retention time prediction of any new solute. The applicability of this tool is also restricted to the IC phases provided by a certain manufacturer. While the ion-exchange retention mechanism for small inorganic ions is governed by electrostatic interactions, hydrophobic interaction has also been shown to contribute to the retention of larger organic species, resulting in deviation of equation 2 from linearity (21). Meanwhile, it has also been shown that the hydrophobic and electrostatic interactions operate independently, which enables the hydrophobic effect to be minimized or even eliminated by the addition of a constant percentage of an organic modifier, such as methanol, to the eluent. Following such modification of the eluent, the LSS retention model based solely on electrostatic interactions has still been found to be applicable and has been used to accurately model retention of larger organic species over varied eluent concentrations (21,22).
In the context of QSRR, IC can also benefit from the structure-based retention prediction approach. The a and b values in equation 2 can serve as dependent variables to correlate with molecular descriptors that are relevant to the ionic species. As an example, Figure 4 illustrates the correlations obtained between experimentally determined and predicted a and b values for a dataset containing 68 inorganic and small organic anions. Separation was performed isocratically using a 250 mm x 4 mm IonPac AS20 column from Thermo Fisher Scientific (formerly Dionex) and 35 mM potassium hydroxide as the eluent. In total, 288 descriptors were calculated by Dragon from the anionic structure of species. A GA-MLR approach was used to select the most significant descriptors from their correlations with the experimental a and b values. Individual MLR models were then derived and used for predicting the a and b values. The optimum number of descriptors in each model (model size) was determined as 30 for the a value and 25 for the b value using cross-validation. Ultimately, the retention time of each ion was calculated by fitting the predicted a and b values in equation 2. Figure 4c illustrates a strong correlation between predicted and experimental retention times with 0.95 as the goodness of fit and MAE better than 0.8 min.


Figure 4: Correlations obtained between experimental and predicted (a) a values and (b) b values as well as (c) experimental retention times and retention times calculated from predicted a and b values for the IC dataset. GA-MLR was used for descriptor selection and modeling.
Further improvement in prediction accuracy is expected by modeling based upon different training sets for different type of ionic species, which could also be combined with the federation of local models approach. Predicted a and b values for any given solute can then be substituted into equation 2 to obtain the predicted retention times at any concentration of eluent. Unlike the current approach, which has been based on columns from one manufacturer, this QSRR methodology will be applicable to any type of IC column for which suitable isocratic retention data are available or feasible to be acquired. It was also shown previously that once available, isocratic IC retention data can be used to predict retention under more-complex elution profiles comprising multiple isocratic and gradient steps, even using columns from different production batches than those used to collect the original isocratic data (18). Additionally, by incorporating a simple algorithm for estimating peak width under complex elution conditions, chromatograms were simulated with a high level of reliability (18). A comprehensive QSRR study in IC can therefore benefit from all of these advantages and come up with a better solution with broader scope and more utility in IC method development.
Method Development in HILIC
HILIC has attracted increasing attention in recent years, particularly because of its suitability in providing complementary separation selectivities to those of reversed-phase LC and IC for the separation of polar and ionized compounds (23). However, unlike reversed-phase LC and IC, the retention mechanism in HILIC is not fully understood, but evidently it includes contributions from partitioning between a water-rich layer adsorbed on the surface of the stationary phase and the bulk mobile phase, adsorption through hydrogen bonding or other dipole-dipole interactions, and electrostatic interactions (23–25). Although HILIC has been infrequently addressed in modeling studies, QSRR models developed so far have been mostly descriptive (rather than predictive), aiming to provide more insight into the retention mechanism of this technique (see, for example, reference 26 and references within) or limited in scope by investigating a very specific set of compounds and using only a limited number of stationary-phase and mobile-phase conditions (26–28). More recently, a hydrophilic-subtraction model has been proposed for the purpose of characterizing HILIC columns, by mimicking the same approach used by Snyder and colleagues in developing their successful hydrophobic-subtraction model for reversed-phase LC (29). Although the proposed model seems to be representative enough for its intended purpose, the same cannot be inferred regarding its suitability for retention time prediction in HILIC, because of the lack of relevant data.
Thus, it appears that a predictive QSRR model fulfilling the industrial requirements for HILIC method development is still lacking. To cover the HILIC knowledge space as much as possible, such a study could be performed in the framework of an experimental design by including the separation parameters having the greatest influence on retention under HILIC conditions. It is evident from a number of studies that such parameters include the stationary phase, the percentage of organic solvent (usually acetonitrile) in the mobile phase, and the pH and concentration of buffer (23). Figure 5 shows a three-dimensional (3D) representation of a typical full-factorial screening design, which can cover the majority of the HILIC knowledge space. To include the effect of stationary phase, a number of representative HILIC columns could be screened under conditions specified by the experimental design. Such a design requires at least nine experiments under different mobile-phase conditions for each column to collect the required retention data of the compounds included in the training set. These data can then be used to establish individual QSRR models for each mobile-phase condition (nine models in total for each stationary phase), which can be subsequently used for predicting the retention times of new compounds. Such a screening study would assist in systematic identification of a suitable starting point for method development in HILIC. Further optimization of the separation can then be performed either by including more experiments in the screening design or by establishing a dedicated optimization design.


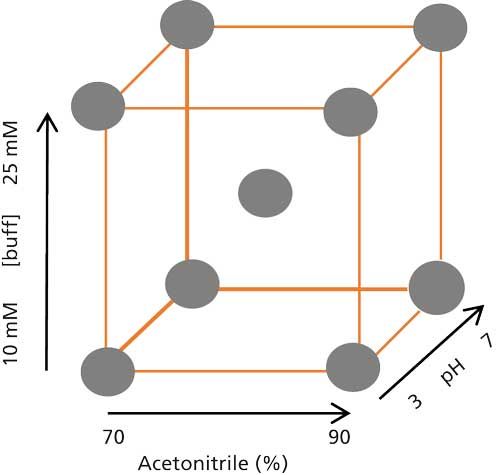
Figure 5: 3D overview of a full-factorial screening design for HILIC, with the percentage of acetonitrile in the mobile phase, and buffer concentration and pH as variables. Each variable is changed at two levels making nine possible combinations (experimental conditions).
Although limited by the availability of time and resources, a comprehensive QSRR study in HILIC also can benefit from a sufficiently large training set. The training set would need to include clusters of compounds with adequate similarity, if the use of approaches such as the federation of local models is desired for improving the prediction accuracy of the models.
Further Considerations
Following the establishment of a workflow for covering the knowledge space of each individual LC technique, the use of strategies for increasing the prediction accuracy of resultant models must be considered. One approach is the localization of predictive models. A locally predictive model is built for each analyte for a particular combination of separation technique, stationary phase, and mobile phase using a training set of compounds having adequate structural similarity to the analyte. As discussed earlier and based on the premise that structural similarity reflects similarity in chromatographic behavior, greater prediction accuracy can be expected for a new compound that is structurally similar to the training set (30). Depending on the availability of resources, many such local models can be created using different training sets and then put together as a directory. The structure of a new compound can be searched against all compounds in the directory to derive the most suitable local model for prediction. This approach is somewhat different from the above-mentioned federation of local models, where instead of using a directory of some prebuilt models, modeling is performed iteratively for each new compound by utilizing only a fraction of (similar) compounds in the retention databases. Although different in strategy, both approaches are expected to provide better prediction accuracies than using a global model for any given compound.
The success of any modeling approach based on structural similarity depends directly on the size of the database and the availability of clusters of similar compounds. This database needs to be both large enough and structurally diverse enough, and ideally should contain sufficient similar compounds to any target compound to allow statistically significant modeling to occur.
There are a number of different approaches to structural similarity, including Tanimoto, Dice, cosine, Hamming distance, and Euclidean distance (6,31). Although these approaches rank structural similarity differently, overall ranking of structures tends to be very similar and none of them seems to have any proven superiority over the others (6). Nevertheless, the Tanimoto approach appears to be the method of choice in computing the fingerprint-based similarity (31).
Predictive models can be established from a small set of preselected molecular descriptors, which are often well-understood physicochemical properties. The predictive quality of models developed using this approach depends strongly on a priori knowledge about the retention mechanism of the chromatographic method under study and also on the availability of relevant descriptors with strong correlation to that retention mechanism. Alternatively, a suitable variable selection method can be used to extract the most predictive molecular descriptors from a large pool of descriptors generated using the available molecular modeling tools. In this approach the prediction accuracy is not limited to a predefined subset of descriptors, and therefore depending on the sample in hand a different subset of descriptors might be found for each analyte as the most relevant for subsequent use in modeling. It is worth mentioning that, considering the prediction accuracy of the final model is of the outmost importance here, the selected descriptors do not necessarily need to be physically meaningful, as would be expected from descriptive models. Clearly, the predictive power of the models resulting from this approach depends primarily on the efficiency of the variable selection method used. GA, for example, is a powerful variable selection method that, in combination with both MLR and PLS, has been used extensively in QSRR studies (32). One popular implementation of GA-PLS in Matlab (The Mathworks Inc.) also features an early stopping approach to prevent the GA from overfitting (9).
Concluding Remarks
The conventional trial-and-error approach in pharmaceutical method development is costly and time- and resource-intensive. Retention time prediction from molecular structure has been proven to be a viable alternative for streamlining the method development activities in industry. To this end, the proposed workflow needs to be comprehensive enough in projecting the chromatographic knowledge space as much as possible. In the pharmaceutical industry the majority of chromatographic knowledge space is covered by reversed-phase LC, IC, and HILIC as the three major techniques. Although the aim is rapid prediction of retention time, the approach taken in achieving this goal can be different depending on the extent of knowledge available for each technique.
In reversed-phase LC, a comprehensive retention prediction scheme can greatly benefit from the availability of the column coefficients for a large fraction of all commercial phases in Snyder’s hydrophobic subtraction model. Accordingly, retention time with adequate accuracy for potentially any given solute can be obtained by pairing column coefficients with their complementary solute coefficients. Therefore, solute coefficients can be the subject of modeling for their subsequent use in retention prediction.
In IC, a and b values in the LSS model are available for an appreciable number of phases and solutes. After they are correlated with solute structures, predicted a and b values can be used for subsequent prediction of retention on available phases. The scope of retention prediction on this technique can be broadened by including more-diverse solutes and also columns from other manufacturers in modeling.
Unlike reversed-phase LC and IC, understanding of the HILIC retention mechanism is not yet mature enough to form the basis of QSRR modeling based on mechanism. Therefore it seems that direct correlation of experimental retention time to molecular descriptors is currently the only available option for retention time prediction in HILIC. A prediction scheme with broad scope can be combined with a suitable experimental design to account for all the major contributors to the HILIC retention mechanism-that is, the stationary phase, the content of organic modifier in the mobile phase, and the pH and concentration of the buffer. Studies have already been performed for classification of HILIC phases, therefore one candidate from each class can be selected for modeling under the conditions defined by the experimental design. Such an approach can reasonably cover the HILIC knowledge space in the quest for a starting point for method development based solely on predicted retention times.
References
- G.L. Reid, G. Cheng, D.T. Fortin, J.W. Harwood, J.E. Morgado, J. Wang, and G. Xue, J. Liq. Chromatogr. Related Technol. 36, 2612–2638 (2013).
- R. Kaliszan, Chromatographia10, 529–531 (1977).
- R. Kaliszan and H. Foks, Chromatographia10, 346–349 (1977).
- Y. Michotte and D.L. Massart, J. Pharm. Sci.66, 1630–1632 (1977).
- S. Schefzick, C. Kibbey, and M.P. Bradley, J. Comb. Chem. 6, 916–927 (2004).
- Y.V. Kazakevich, M. McBrien, and R. LoBrutto, in HPLC for Pharmaceutical Scientists, Y.V. Kazakevich and R. LoBrutto, Eds. (John Wiley & Sons, Inc., Hoboken, New Jersey, 2007).
- T. Baczek and R. Kaliszan, J. Chromatogr. A987, 29–37 (2003).
- K. Baumann, TrAC, Trends Anal. Chem.22, 395–406 (2003).
- R. Leardi and A. Lupiáñez González, Chemometr. Intell. Lab. Syst.41, 195–207 (1998).
- M. Talebi, G. Schuster, R.A. Shellie, R. Szucs, and P.R. Haddad, J. Chromatogr. A1424, 69–76 (2015).
- T.H. Miller, A. Musenga, D.A. Cowan, and L.P. Barron, Anal. Chem.85, 10330–10337 (2013).
- N.S. Wilson, M.D. Nelson, J.W. Dolan, L.R. Snyder, R.G. Wolcott, and P.W. Carr, J. Chromatogr. A961, 171–193 (2002).
- L.R. Snyder, J.W. Dolan, and P.W. Carr, Anal. Chem.79, 3254–3262 (2007).
- Y. Zhang and P.W. Carr, J. Chromatogr. A1216, 6685–6694 (2009).
- http://www.usp.org/app/USPNF/columnsDB.html.
- M.-J. Rocheleau, Curr. Pharm. Anal. 4, 25–32 (2008).
- P.R. Haddad and P.E. Jackson, Ion Chromatography: Principles and Applications (Elsevier, 1990).
- R.A. Shellie, B.K. Ng, G.W. Dicinoski, S.D.H. Poynter, J.W. O’Reilly, C.A. Pohl, and P.R. Haddad, Anal. Chem.80, 2474–2482 (2008).
- B.K. Ng, G.W. Dicinoski, R.A. Shellie, and P.R. Haddad, “Advances in Ion Chromatography” supplement to LCGC North Am. 31(4b), 33–37 (2013).
- S.H. Park, R.A. Shellie, G.W. Dicinoski, G. Schuster, M. Talebi, P.R. Haddad, R. Szucs, J.W. Dolan, and C.A. Pohl, J. Chromatogr. A1436, 59–63 (2016).
- P. Zakaria, G.W. Dicinoski, B.K. Ng, R.A. Shellie, M. Hanna-Brown, and P.R. Haddad, J. Chromatogr. A1216, 6600–6610 (2009).
- P. Zakaria, G. Dicinoski, M. Hanna-Brown, and P.R. Haddad, J. Chromatogr. A1217, 6069–6076 (2010).
- A. Kumar, J.C. Heaton, and D.V. McCalley, J. Chromatogr. A1276, 33–46 (2013).
- W. Bicker, J. Wu, M. Lammerhofer, and W. Lindner, J. Sep. Sci.31, 2971–2987 (2008).
- E. Tyteca, A. Periat, S. Rudaz, G. Desmet, and D. Guillarme, J. Chromatogr. A1366, 136–136 (2014).
- R.I. Chirita, C. West, S. Zubrzycki, A.L. Finaru, and C. Elfakir, J. Chromatogr. A1218, 5939–5963 (2011).
- N.S. Quiming, N.L. Denola, Y. Saito, and K. Jinno, Anal. Bioanal. Chem. 388, 1693–1706 (2007).
- N.S. Quiming, N.L. Denola, Y. Saito, and K. Jinno, J. Sep. Sci.31, 1550–1563 (2008).
- J. Wang, Z. Guo, A. Shen, L. Yu, Y. Xiao, X. Xue, X. Zhang, and X. Liang, J. Chromatogr. A1398, 29–46 (2015).
- A. Tropsha, Mol. Inform.29, 476–488 (2010).
- P. Willett, Drug Discovery Today11, 1046–1053 (2006).
- M. Goodarzi, Y.V. Heyden, and S. Funar-Timofei, TrAC, Trends Anal. Chem. 42, 49–63 (2013).
Mohammad Talebi, Soo Hyun Park, Maryam Taraji, Yabin Wen, Ruth I.J. Amos, and Paul R. Haddad are with the Australian Centre for Research on Separation Science (ACROSS), at the University of Tasmania in Australia. Robert A. Shellie is with Trajan Scientific Australia in Melbourne, Australia. Roman Szucs is with Pfizer Global Research and Development in Sandwich, UK. Christopher A. Pohl is with Thermo Fisher Scientific in Sunnyvale, California. John W. Dolan is with LC Resources in McMinnville, Oregon. Direct correspondence to: mtalebi@utas.edu.au







HILIC Peptide Retention Times Predicted Using New Approach
October 29th 2024Manitoba Centre for Proteomics and Systems Biology scientists produced a new means of predicting peptide retention times for hydrophilic interaction liquid chromatography (HILIC) at acidic pH in formic-acid based eluents.



Systematic Evaluation of HILIC Stationary Phases for MS Characterization of Oligonucleotides
Hydrophilic interaction chromatography–mass spectrometry (HILIC-MS) offers a flexible and efficient alternative to ion-pairing reversed-phase liquid chromatography (IP-RPLC) for oligonucleotide analysis, with column selectivity and mobile phase pH being key factors in optimizing retention and detection.


