Headspace Solid-Phase Microextraction Coupled with Gas Chromatography–Mass Spectrometry (HS-SPME-GC–MS) for the Characterization of Cigar Leaves
Manufacturers and consumers are increasingly concerned about high-quality cigar products with clear geographical sources. To characterize the chemical profiles of different geographical origins of cigar leaf samples, headspace solid phase microextraction coupled with gas chromatography–mass spectrometry (HS-SPME-GC–MS) combined with a chemometrics method was used in this study. The chromatography and chemical composition of cigar leaf samples from 16 geographical origins were acquired and profiled. The chemical markers contributing to the differentiation of cigar leaf samples were observed and characterized by a supervised orthogonal projections to latent structures discriminant analysis (OPLS-DA) method of chemometrics. Eight chemical markers were tentatively identified to be used as specific chemical markers for the differentiation of geographical cigar leaf samples. HS-SPME-GC–MS method coupled with chemometrics analysis has potential to be used for discriminating different geographical origins for cigar leaf samples.
The tobacco plant (Nicotiana tabacum) is widely planted and consumed all over the world. By manipulating its leaves, different products can be prepared for smoking or chewing. Today, cigars and rope tobacco (tobacco mincing and rolling) are popular.
The quality of smoke produced in the normal combustion process of cigars depends largely on the chemical composition of tobacco (1–3). The chemical composition of tobacco is affected by many factors, among which the origin of the tobacco is the most important. To obtain the best quality cigar tobacco, we must recognize the source of cigar tobacco. Cuba, Indonesia, Dominica, Brazil, Nicaragua, and China all have famous cigar leaf planting locations.
Nowadays, manufacturers and consumers are increasingly concerned about high-quality cigar products with clear geographical sources. Flavor is one of the important distinguishing feature of cigar quality and consumer affection. The taste of a cigar is very complex, depending on the compounds it contains, such as ketones, esters, alcohols, nitrogen compounds, acids, aldehydes, and lactones. More than 1000 aromatic compounds have been funded, covering a wide range of polarity and volatility (4–7).
Analytical techniques and separation techniques, such as high performance liquid chromatography (HPLC) and matrix assisted laser desorption ionization time of flight mass spectrometry (MALDI–TOF-MS), have been used to determine the geographic origin of food (8–10). This multifactor method requires chemometric analysis of the data provided by analytical instruments.
Headspace solid phase microextraction (HS-SPME) is particularly beneficial for volatile and “semi-volatile” substances because of the low chromatographic background. This method is especially suitable for the detection of volatile and flavor substances. Taking vegetable oil as an example, Yang and Peppard (11) introduced flavor analysis of juice drinks and butter. SPME was used to analyze the varietal characteristics by using different types of fibers. There are many types of SPME fibers, which can be used to analyze many kinds of compounds. Among the existing SPME fibers, SPME fibers containing liquid and solid components have high sensitivity. The SPME program is simple to use, with a duration of less than 1 h without solvent extraction. This method is also cheaper and allows the feature of headspace analysis, making it an optimized method for analyzing aroma in cigar leaves (12–14).
Traditional methods to separate aroma and flavor from tobacco, such as solvent extraction, adsorbent capture, simultaneous distillation extraction, or the combination of these methods, is labor-intensive because of the very low concentration of relevant compounds in a complex matrix, and often the sensitivity is not sufficient (15,16).
Vas and Vekey (17) recently published a comprehensive review on the subject of SPME. HS-SPME is particularly suitable for the analysis of tobacco leaf components because most of these compounds are volatile and can be adsorbed by SPME fiber materials. Therefore, this technology has been used to analyze tobacco constituents many times. In most cases, these methods have been limited to substances such as organic acids like alkenylbenzene (18–22).
HS-SPME and gas chromatography (GC) are the most suitable techniques for determining the origins of tobacco leaves used for cigar-production. In this study, a quick and simple method of cigar leaf screening is introduced, and some of the cigar leaves are quantitatively analyzed by HS-SPME-GC–MS before principal component analysis (PCA) and orthogonal projections to latent structures discriminant analysis (OPLS-DA) is used to analyze the aroma spectrum of 16 cigar leaves.
Materials and Methods
Reference Substances and Reagents
Phenylethyl acetate, which served as the internal standard, was obtained from Sigma–Aldrich. The chemicals and reagents used in analytical grade purity were obtained from Merck; ultrapure water was purified by a Milli-Q system.
Cigar Leaf Samples
For this study, 16 species of cigar leaf samples were collected from five countries. Packs of 16 different common cigar leaves were provided by the Sichuan tobacco company (Sichuan, China), the information of which is displayed in Table I. The 3R4F reference cigarette was purchased from the University of Kentucky. The cigar leaf samples were stored at −40°C prior to analysis.


Instruments and Accessories For GC–MS and HS-SPME
An Agilent gas chromatograph coupled to a mass selective detector (MSD) (7890- 5977B) was used for the GC–MS analysis. A multipurpose sampler called MPS2 (Gerstel) for automatic performance of the HS-SPME measurements was also used.
A DB-5MS capillary column (30 m × 0.25 mm × 0.25 μm) from Agilent was used for the gas chromatographic analysis with helium as the carrier gas at a flow rate of 1.0 mL/min.
The SPME fibers tested in this work were 100 μm of polydimethyl-siloxane (PDMS), 85 μm carboxen/polydimethylsiloxane (CAR/PDMS), 65 μm polydimethylsiloxane/divinylbenzene (PDMS/DVB), and 50 μm divinylbenzene/carboxen/polydimethylsiloxane (DVB/CAR/PDMS). All SPME fibers were purchased from Supelco.
The optimal analyses were performed with the 50 μm DVB/CAR/PDMS fiber. All GC–MS analysis were performed using the enhanced ChemStation G1701 DA, version F.01.01.2317 software from Agilent Technologies.
Sample Preparation For Qualitative Screening Method and HS-SPME
In the optimized qualitative screening method, each cigar sample was analyzed under three different conditions. An 0.1 g sample of the cigar leaf was placed in a 22 mL headspace vial. Then, 50 μL of the phenylethyl acetate methanol solvent with a concentration of 100 μg/mL was added as the internal standard. Then, the 22 mL headspace vials were tightly closed and placed into the vial rack of the sampler. The samples were extracted for 40 min at 80 °C and desorpted at 280 °C for 3 min.
Gas Chromatography–Mass Spectrometry (GC–MS)
Using helium as the carrier gas, the flow rate was 1.0 mL/min, and the injection mode was splitless at 280 °C. The optimized method was as follows: 2 min at 60 °C, then the temperature was increased 10 °C/min up to 280 °C, and then it remained for 10 min at 280 °C. The temperature of quadrupole and the ion source were 150 °C and 230 °C, respectively. Analytes were detected in scan mode from m/z = 31 to 550. Peak recognition in scanning mode was performed using the software MS Spectral Library NIST17 (National Institute of Standards and Technology).
Qualitative and Quantitative Analyses
By comparing the retention time and mass spectra of the samples with the mass spectra present in the National Institute of Standards and Technology (NIST) MS 17 spectral database, the compounds were qualitative. Quantitative analysis was carried out by the internal standard method.
Statistical Analyses
All data of the peaks were exported into the Simca software (v. 13.0) for OPLS-DA analysis. One-way analysis of variance (ANOVA) was used to analyze the significant difference of markers in different samples. The differences were considered statistically significant with the analysis of variance, P ≤ 0.05, maximum folding change ≥2, and the valuable importance in the projection (VIP) value was >1 as the limiting conditions.
Results
HS-SPME-GC–MS Optimization
Volatiles present in cigar leaves were extracted using a 50 μm DVB/CAR/PDMS fiber at 80 °C for 40 min. For this study, 95 compounds that were identified and quantified are presented. Because of the different structure, polarity, and volatility of the target compounds, a medium polarity fiber material was needed. Four fibers (PDMS, PDMS/DVB, CAR/PDMS, DVB/CAR/PDMS) were compared for extracting volatile compounds. The experiment was performed using a China cigar leaf that contained the volatile compounds of esters, acids, and alcohols (23–25). The performance of each fiber was compared by the number of compounds identified and the response area, in which a group of 50 volatile compounds were considered (25 esters, 14 terpenes, 5 alcohols, and 6 acids).
Compared with other fibers, the extraction rate of DVB/CAR/PDMS and PDMS/DVB coatings is higher for volatiles. The results of the DVB/CAR/PDMS fibers were significantly different from the other three fibers, and showed that the total area of esters, alcohols, and acids was the highest, and that more volatile compounds (95) were detected in these functional groups. In addition, the repeatability of DVB/CAR/PDMS was less than 14%. The extraction efficiency of benzyl alcohol, pyridine, and benzaldehyde was slightly better than that of PDMS/DVB fiber, as shown in Table II. This is the most commonly used fiber in the literature (26–29).


Although DVB/CAR/PDMS and PDMS/DVB are bipolar fibers covered with porous solid coating, which indicates that the extraction of analytes is carried out by adsorption, the qualitative and quantitative behavior of PDMS/DVB fiber is worse than that of DVB/CAR/PDMS, and 63 compounds are detected. The extraction efficiency of the PDMS/DVB fiber for neophytadiene, one of the most important compounds in cigar leaves, is also very low. This choice could be confirmed by comparing the peak areas of alkaline, neutral, and acidic compounds in SIM mode.
Nonpolar-coating PDMS fibers could only identify 39 volatile compounds. However, the peak area of these compounds was low, which indicates that PDMS fibers are not suitable for quantitative analysis. The combination of DVB and CAR increases the porosity distribution and polarity of the fiber while improving the retention rate of analyte on the fiber compared with the coating only composed of PDMS.
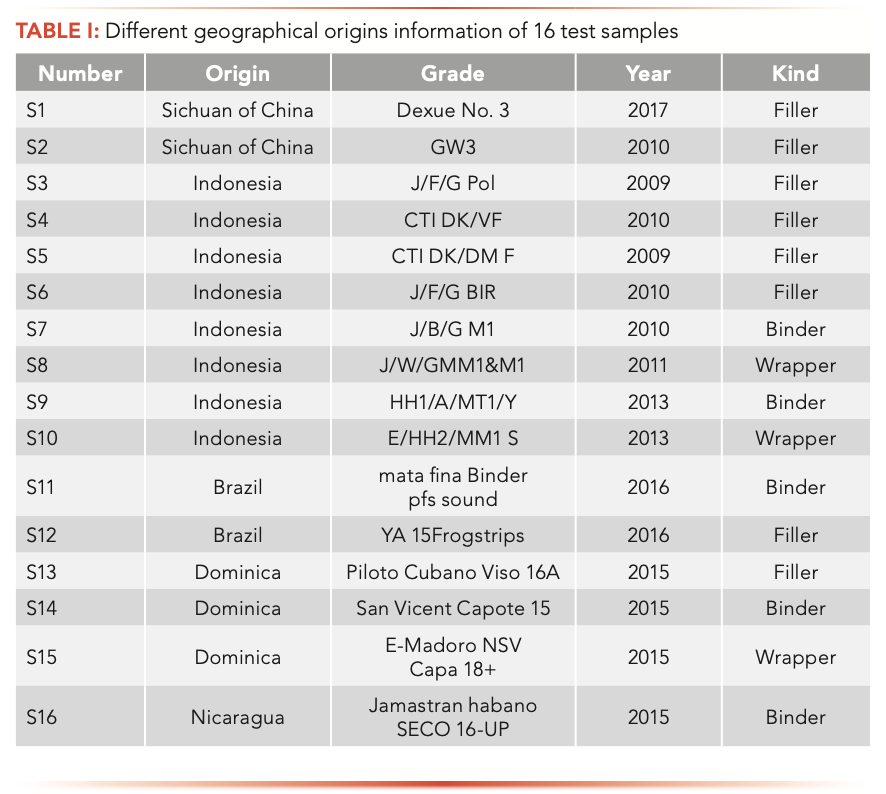
The extraction efficiency of DVB/CAR/PDMS fiber for volatile components from cigar leaves were the highest, and the repeatability was better (RSD 10%). Previous studies indicated that the DVB/CAR/PDMS fiber is the most suitable because of its extraction ability for compounds (molecular weight 40–275) (30–34). As an example, the total ion chromatograms obtained after basic preparation are shown in Figure 1.


FIGURE 1: Total ion chromatograms of a cigar sample measured by HS-SPME and GC–MS with the 50 μm DVB/CAR/PDMS SPME fiber after basic sample preparation.
OPLS-DA Analysis
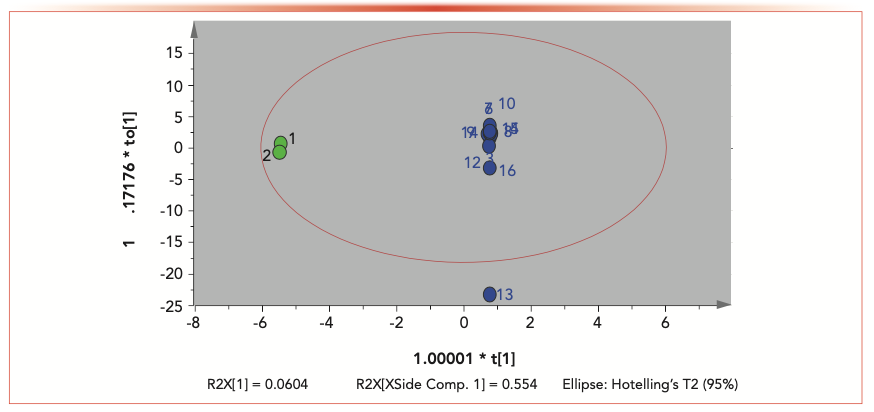
All results of 16 cigar leaf samples were exported into the Simca software (v 13.0) for OPLS-DA analysis. Outliers and classification trends of 16 cigar leaf samples were observed in the OPLS-DA results (Figure 2). The score plot obtained by OPLS-DA showed that there was a clear distinguish between the S1 and S2 groups and the S3 to S16 group, indicating that the cigar leaf sample from China Sichuan was very different from other samples in Indonesia, Dominica, Brazil, and Nicaragua. Cigar leaf samples from China Sichuan (S1 and S2 groups) clustered together and separated from other samples in Indonesia, Dominica, Brazil, and Nicaragua (Figure 3). R2X of the OPLS-DA model (0.554) showed that the model had good adaptability and predictability. S1 and S2 samples from China Sichuan province clustered together respectively, which shows that there are significant differences between domestic and foreign samples; the separation between S3–S10, S11 and S12, S13–S15, and S16 indicated that samples in Indonesia, Dominica, Brazil, and Nicaragua also had significant differences.

 scores plot of 16 cigar samples from different geographical origins: (CI green) domestic samples from China; FO (blue) foreign samples from outside of China.")
FIGURE 2: Orthogonal partial least squares discriminant analysis (OPLS-DA) scores plot of 16 cigar samples from different geographical origins: (CI green) domestic samples from China; FO (blue) foreign samples from outside of China.

, domestic samples from China; and group 2 (blue), foreign samples from outside of China.")
FIGURE 3: Clustering of 16 samples showed that there are significant differences between group 1 (green), domestic samples from China; and group 2 (blue), foreign samples from outside of China.
Identification of Chemical Markers
Based on the retention behavior and mass distribution, potential chemical markers in cigar leaves from different geographical origins were identified. First, the OPLS-DA model was constructed from the Simca software because potential biomarkers of interest can be extracted from loading plots and VIP plots of that model.
In addition, with the analysis of variance of P ≤ 0.05, maximum folding change of ≥2, and the VIP value >1 as the limiting conditions, the compounds with significant changes were screened out to reduce the “false discovery rate (FDR)”.
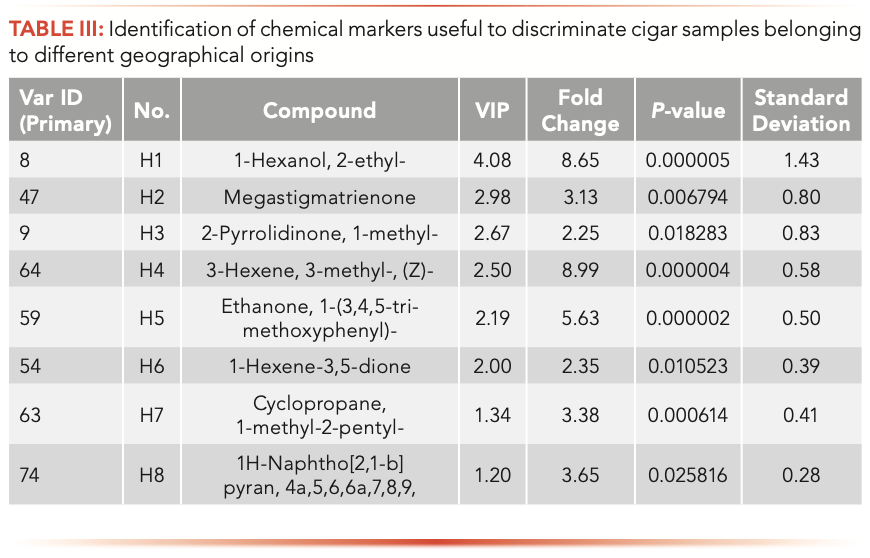
According to the condition detailed above, eight chemical markers (1-hexanol, 2-ethyl-; megastigmatrienone; 2-pyrrolidinone, 1-methyl-; 3-hexene, 3-methyl-, (Z)-; ethanone,1-(3,4,5-trimethoxyphenyl)-; 1-hexene- 3,5-dione; cyclopropane, 1-methyl- 2-pentyl-; and 1H-naphtho[2,1-b] pyran, 4a,5,6,6a,7,8,9; short named H1–H8) in cigar leafs from different origins were identified (Table III).


Six repeated QC samples were analyzed to evaluate the repeatability of SPME-GC–MS. The relative standard deviations of peak areas and retention time were 2.3–5.7 and 0–0.14%, respectively. Quality control (QC) samples were kept in the automatic sampler for 5, 10, 20, and 40 h to evaluate the stability of the instrument. The relative error of peak area is less than 10.5%, which has good repeatability and stability.
To more clearly describe the differences of chemical markers in samples from different geographical origins, the relative intensity of chemical markers are shown in Table III and Figure 4. The max fold changes of H1–H8 were 8.99, 8.64, 5.63, 3.64, 3.38, 3.13, 2.34, and 2.25, respectively. The results of Figure 4 and Table II showed that the content of H1 in the 16 samples had the biggest difference, and the content of H1 in S1 was the highest, which was about 45.08 more times than S10; the content of H5 in S1 and S2 were similar, which was higher than other 14 samples; and the content of H2 and H3 in all samples were similar.

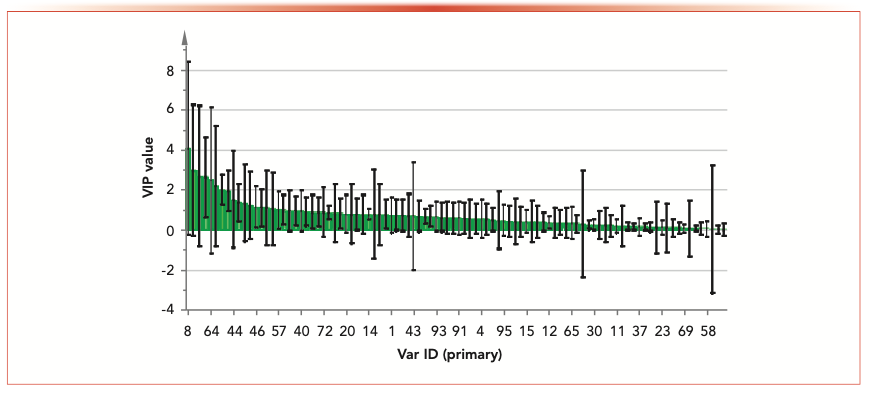
 showing the variable importance in the projection (VIP) value of chemical markers.")
FIGURE 4: Orthogonal projections to latent structures discriminant analysis (OPLS-DA) showing the variable importance in the projection (VIP) value of chemical markers.
The content of H6 in S16 was the highest; the content of S2 and S13 were higher than S1, S3–S12, S14, and S15. The content of H7 in S1 and S2 was similar, which was higher than the other 14 samples.
Discussion
Manufacturers and consumers are increasingly focused on high-quality cigar products with well-defined geographical origins. Therefore, the research of the source of cigar leaves is an important subject. Flavor is one of the important indicators of cigar quality and consumer acceptance. In this study, 16 cigar leaf samples were collected from five countries. Eight representative chemical markers related to the differences were made on 16 cigar leaf samples. The contents of H1 and H5 were the highest in S1 and S2. The results showed that H1 and H5 could be used as specific chemical markers of S1 and S2; H3 could be used as specific chemical markers of S16; H4 and H6 could be used as specific chemical markers of S6–S8; and H8 could be used as a unique chemical marker of S13.
The OPLS-DA results of different samples showed that the cigar leaf from the Sichuan provinces in China were different from the foreign samples, indicating that the China samples from the Sichuan province had some similarity. The samples from Indonesia, Dominica, Brazil, and Nicaragua were also clustered together, respectively.
Cigar leaves contain a variety of alcohols, terpenes, ketones, esters, acids, aldehydes. 1-hexanol 2-ethyl-(H1) contains a slight floral fragrance, which increases the floral scent when smoking cigars.
Megastigmatrienone (H2) is a key flavor compound in tobacco. As the most abundant substance in neutral aroma components, megastigmatrienone was found and its chemical structure was identified. Scientists also found that burley tobacco concentrate contains nearly 10% of the megastigmatrienone chemicals, and it exists more or less in flue-cured tobacco. It is a degradation product of carotenoids and formed by the degradation of lutein, which has a very important contribution to the aroma of tobacco leaves.
The 16 cigar leaf samples from different geographical origins were identified by HS-SPME-GC–MS and chemometrics. These chemical markers with significant aroma can be used to discriminate the geographical origin of cigar leaf samples. Chemometrics technology has potential application prospects in the discovery and quality evaluation of flavor components in different geographical environment, which helps us to find natural substitutes for geographical origins more quickly.
Conclusion
Classification and differentiation of cigar leaves according to their producing areas is an important basis to ensure the quality of tobacco leaves. The chemical constituents of cigar leaves from different areas were characterized by HS-SPME-GC–MS. HS-SPME combined with GC–MS is a fast, simple and solvent-free method. The chromatographic results of adsorption with PDMS, PDMS/DVB, DVB/CAR/PDMS, and CAR/PDMS coated fibers showed that DVB/CAR/PDMS was the best one for analyzing alcohols, esters and acids in cigar leaves.
The OPLS-DA method was used to distinguish the cigar leaf samples from different geographical origins. The marker compounds used to differentiate cigar leaves were discovered. There were eight chemical markers that were preliminarily identified and semi-quantitated in cigar leaf samples from several countries. The chemical markers results showed that there were significant differences between different regions. These chemical markers can be used to distinguish the origin of samples from different countries. The results show that chemometrics technology may be used to find aroma components related to complex components and different geographical environments of cigar leaves, which will help us find natural substances for geographical sources quicker.
Funding Sources
The study was supported by Key Laboratory of Chinese cigar fermentation of China Tobacco Sichuan Industrial Co., Ltd. (ctx201901).
References
(1) J.K. Ha and R.C Lindsay, Flavour Fragrance J. 6, 81–85 (1991).
(2) Z.Y. Chen, J.L. Tan, G.Y. Yang, M.M. Miao, Z.Y. Chen, and T.F. Li, Phytochem. Lett. 5, 233–235 (2012).
(3) S. Yan and G.S. Liu, Phytochem. Lett. 36, 42–48 (2020).
(4) E.L. Carmines, Food Chem. Toxicol. 40(1), 77–91 (2002).
(5) J.D. Heck, C.L. Gaworski, N. Rajendran, and R.L. Morrissey, Inhal. Toxicol. 14, 1135–1152 (2002).
(6) E. Roemer, R. Stabbert, K. Rustemeier, D.J. Veltel, T.J. Meisgen, W. Reininghaus, R.A. Carchman, C.L. Gaworski, and K.F. Podraza, Toxicology 195, 31–52 (2004).
(7) K. Rustemeier, R. Stabbert, H.J. Haussmann, E. Roemer, and E.L. Carmines, Food. Chem. Toxicol. 40(1), 93–104 (2002).
(8) M.L. Chen, W.Q. Chang, J.L. Zhou, Y.H. Yin, W.R. Xia, J.Q. Liu, L.F. Liu, and G.Z. Xin, J. Pharmaceut. Biomed. 145, 666–674 (2017).
(9) S. Liu, Y.Z. Liang, and H.T. Liu, J. Chromatogr. B 1015–1016, 82–91 (2016).
(10) R.S. Schwartz, I.T. Hecking, J. Anal. At. Spectrom 6, 637–642 (1991).
(11) X. Yang and T. Peppard, J. Arg. Food. Chem. 42, 1925–1930 (1994).
(12) P. Guedes de Pinho, R.F. Goncalves, P. Valentao, D.M. Pereira, R.M. Seabra, P.B. Andrade, and M. Sottomayor, J. Pharmac. Biomed. Anal. 49, 674–685 (2009).
(13) E. Carasek and J. Pawliszyn, J. Agric. Food Chem. 54, 8688–8696 (2006).
(14) L. Tat, P. Comuzzom, I. Stolfo, and F. Battistutta, Food Chem. 93, 361–369 (2005).
(15) Z. Wu, W.W. Weeks, and R.C. Long, J. Agric. Food Chem. 40, 1917–1921 (1992).
(16) F. Peng, L. Sheng, B. Liu, H. Tong , and S. Liu, J. Chromat. A. 1040(1), 1–17 (2004).
(17) G. Vas and K. Vekey, J. Mass Spectrom. 39, 233–254 (2004).
(18) P. Kusch, LCGC North Am. 36(1), 52–61 (2018).
(19) S.B. Stanfill, A.M. Calafat, C.R. Brown, G.M. Polzin, J.M. Chiang, C.H. Watson, and D.L. Ashley, Food Chem. Toxicol. 41, 303–317 (2003).
(20) C.H. Watson and D.L. Ashley, J. Chromatogr. Sci. 38, 137–144 (2000).
(21) S.S. Yang, C.B. Huang, and I. Smetena, J. Chromat. A 942, 33–39 (2002).
(22) Z. Li, L. Wang, G. Yang, H. Shi, C. Jiang, W. Liu, and Y. Zhang, J. Chromatogr. Sci. 41, 36–40 (2003).
(23) R.R. Baker, J.R. Pereira da Silva, and G. Smith, Food Chem. Toxicol. 42(s), S3–S37 (2004).
(24) R.R. Baker, J.R. Pereira da Silva, and G. Smith, Food Chem. Toxicol. 42(s), S39–S52 (2004).
(25) R.R. Baker, E.D. Massey, and G. Smith, Food Chem. Toxicol. 42(s), S53–S83 (2004).
(26) R. Perestrelo, M. Caldeira, F. Rodrigues, and J.S. Camara, J. Sep. Sci. 31, 1841– 1850 (2008).
(27) L. Ferreira, R. Perestrelo, M. Caldeira, and J.S. Camara, J. Sep. Sci. 32, 1875–1888 (2009).
(28) J. Oliveira, I. Araujo, O. Pereira, J. Maia, A. Amaral, and M.O. Maia, Anal. Chim. Acta 513, 269–275 (2004).
(29) J. Oliveira, M. Faria, F. Sa, F. Barros, and I. Araujo, Anal. Chim. Acta 563, 300–309 (2006).
(30) B. Mendes and J. Camara, Talanta 88, 79–94 (2012).
(31) R.M. Pena, J. Barciela, C. Herrero, and S. Garcia-Martin, J. Sci. Food Agric. 85, 1227–1234 (2005).
(32) L. Rebiere, A.C. Clark, L.M. Schmidtke, P.D. Prenzler, and G.R. Scollary, Anal. Chim. Acta 660, 149–157 (2010).
(33) L. Setkova, S. Risticevic, and J. Pawliszyn, J. Chromatogr. A 1147, 224–240 (2007).
(34) A. Zander, A.G. Bishop, and P.D. Prenzler, Anal. Chim. Acta 530(2), 325–333 (2005).
Hongfei Zhang, Fengpeng Zhu, Yongqiang Pang, Yanbo Luo, Xiangyu Li, and Xingyi Jiang are with the China National Tobacco Quality Supervision and Test Center in Zhengzhou, China. Juan Yang, Cheng Luo, Beibei Zhu, and Dongliang Li are with the Technology Center at the China Tobacco Sichuan Industrial Co., Ltd., in Chengdu, China. Direct correspondence to Dongliang Li at: 360188228@qq.com or Hongfei Zhang at: hfzhang1983@126.com.

 for the Characterization of Cigar Leaves")
 and Thin Layer Chromatography (TLC) Densitometry Methods for the Quantification of Stigmasterol 3-O-β-D-Glucopyranoside (S3G) in Balanites Aegyptiaca Extract: Application to Newly Formulated Balanites Capsules")













Identifying and Rectifying the Misuse of Retention Indices in Gas Chromatography
October 31st 2024LCGC International spoke to Phil Marriott and Humberto Bizzo about a recent paper they published identifying the incorrect use of retention indices in gas chromatography and how this problem can be rectified in practice.




")

