Ultra-Sensitive and Rapid Analysis of Per- and Polyfluoroalkyl Substances (PFAS) in Water
A method is described using a triple quadrupole LC–MS instrument with isotopic dilution to obtain the highest accuracy and confidence for analysis of per- and polyfluoroalkyl substances (PFAS) in water. Excellent method spike recoveries and robustness were found in wastewater.
Per- and polyfluoroalkyl substances (PFAS) are chemicals widely used in consumer products because of their unique and desirable chemical properties. Because of widespread usage and environmental persistence, legacy PFAS are ubiquitous in the environment, and new fluorochemicals are being found in the environment frequently. Public interest and concern has increased pressure to develop comprehensive methods for sensitive analysis in different types of water. We discuss a method for 35 PFAS compounds analyzed in nonpotable waters that is sensitive, robust, and precise, and has high-throughput capabilities because of the fast sample preparation that makes it ideal for laboratories looking to run at high capacity. The analyte lists overlap with American Society for Testing and Materials (ASTM) 7979 and Draft Environmental Protection Agency (EPA) 8327, with some additions of emerging PFAS, such as Adona and the components of F53B because of their increasing presence in the environment. Sample preparation was minimal with 1:1 dilution with methanol followed by filtration. Compound separation was achieved on a 1260 liquid chromatograph (LC) and analyzed with an Agilent 6495 triple quadrupole LC–MS instrument. Compounds were quantitated with isotopic dilution to obtain the highest accuracy and confidence. Excellent method spike recoveries and robustness when running hundreds of wastewater samples were seen, proving this methodology is suitable for use in a high-throughput laboratory with high data accuracy and good sensitivity.
Per- and polyfluoroalkyl substances (PFAS) are a group of >4000 man-made substances that have been in use since the 1940s (1). These substances have unique chemical properties because of the carbon–fluorine bond that make their use desirable in a number of industrial and household products. Common applications of PFAS are in products used as fire-fighting foams, nonstick cookware, water repellent clothing and furniture, food-packing materials, and many others. The same properties that make them tough, durable, and resistant to temperature and degradation also mean they can be persistent in the environment (2,3). These substances, particularly those with less than seven carbon-chain lengths, are bioaccumulative and have various adverse health effects in humans and wildlife (4,5).
There are several hundred PFAS that have been detected in the environment and studies have shown their presence in many major water bodies, soil, air, food, and even in polar bears in the arctic. Several studies have also detected PFAS in human blood across the globe (6–9). Although there are several thousand PFAS known to have been created, they can largely be divided in 10 sub-classes. Of these, the most commonly studied classes are perfluorocarboxylic acids (PFCAs) and perfluorosulfonic acids (PFSA). Two PFAS in particular, perfluorooctane sulfonic acid (PFOS) and perfluorooctanoic acid (PFOA), have been widely used in the past and are being considered for regulatory determination globally. These two compounds have been detected frequently in the environment, and there is mounting toxicological data that points to adverse effects in animals and humans (10). As a result, these compounds are designated as persistent organic pollutants (POPs) under the United Nations Stockholm Convention, which is an international treaty aimed at eliminating the use of POPs globally. In 2015, several major manufacturers of these chemicals decided to voluntarily phase out the manufacture and use of PFOA and PFOS in the United States as part of a stewardship program with the U.S. Environmental Protection Agency (USEPA) (11). This decision has led to a change in manufacturing and production of shorter-chain PFAS compounds. Subsequently, we have started to detect these compounds in the environment with hexafluoropropylene oxide dimer acid (HFPO-DA), also commonly referred to by the brand name GenX, a particular PFAS of interest in the research and regulatory community.
Because of their potential adverse effects, frequent detections in the environment, and the resulting public outcry, PFAS compounds, especially PFOA and PFOS, are under review for regulation in water globally. In the United States, the EPA has established a drinking water health advisory level of 70 ng/L for PFOA and PFOS combined and is evaluating setting a drinking water maximum contaminant level (MCL) for these compounds soon. More than 25 states in the United States have implemented their own regulatory limits for PFAS in drinking water, with many being much more stringent than the USEPA’s value and around the single-digit nanogram per liter level. The European Union has listed PFOA and PFOS as priority hazardous substances under its Water Framework Directive (WFD) and several other PFAS are listed under the Registration, Evaluation, Authorization, and Restriction of Chemicals (REACH) regulation to restrict their use and manufacture in commercial and household products. Several European states, such as Germany, Sweden, and Denmark also have individual limits for specific PFAS in water and soil. Many other countries like Japan, Korea, Australia, and others have also set preliminary or final regulatory limits for certain PFAS compounds in water. It is expected, with the increased toxicological information and identification of newer PFAS, that these regulatory lists will only increase in scope and lead to lower regulatory limits globally. It is thus prudent to have sensitive analytical techniques to measure a vast range of legacy and emerging PFAS in water to have adequate baseline monitoring data to assist regulators in setting regulatory guidance and ensuring safe water.
Analysis of PFAS in the environment can be divided into quantification and identification of new PFAS. Identifying new PFAS is best performed using a high-resolution accurate mass spectrometer (MS), such as quadrupole time of flight (QTOF) instruments, that measure molecular masses accurately and then, with the help of sophisticated software platforms, identify chemical structures. One example is the identification of perfluoroether carboxylic acids (PFECAs) like GenX using LC–QTOF by Strynar and others (12). The gold standard for quantifying PFAS at low nano- gram per liter levels with robustness and reliability though is with tandem quadrupole LC–MS/MS instruments (13). Several standard methods for quantification of sub-sets of PFAS in water exist now. The USEPA has released two methods for analysis of PFAS in drinking water. In 2018, EPA method 537.1 was released to analyze 18 PFAS using solid phase extraction (SPE) and LC–MS/MS while EPA method 533, released in December 2019, analyzes 25 PFAS, including short-chain ones using SPE and LC–MS/MS (14,15). Both of these methods were developed for analysis in drinking water only. In 2019, the EPA released a draft of EPA 8327 that analyzes 28 PFAS in non-potable waters (surface water, ground water, and wastewater) using a dilute-and-shoot technique with minimal sample preparation (16). This method allows much faster analysis and turnaround of samples because it does not require SPE, but as a result needs a more sensitive MS/MS instrument. EPA 8327 is similar to American Society for Testing and Materials (ASTM) 797917 that can be used for analysis of PFAS in nonpotable waters as well. The ISO/DIS 21675 is a method developed for analysis of PFAS in drinking water, natural water, and wastewater using SPE and LC–MS/MS as well.
Experimental
Standards and Water Samples
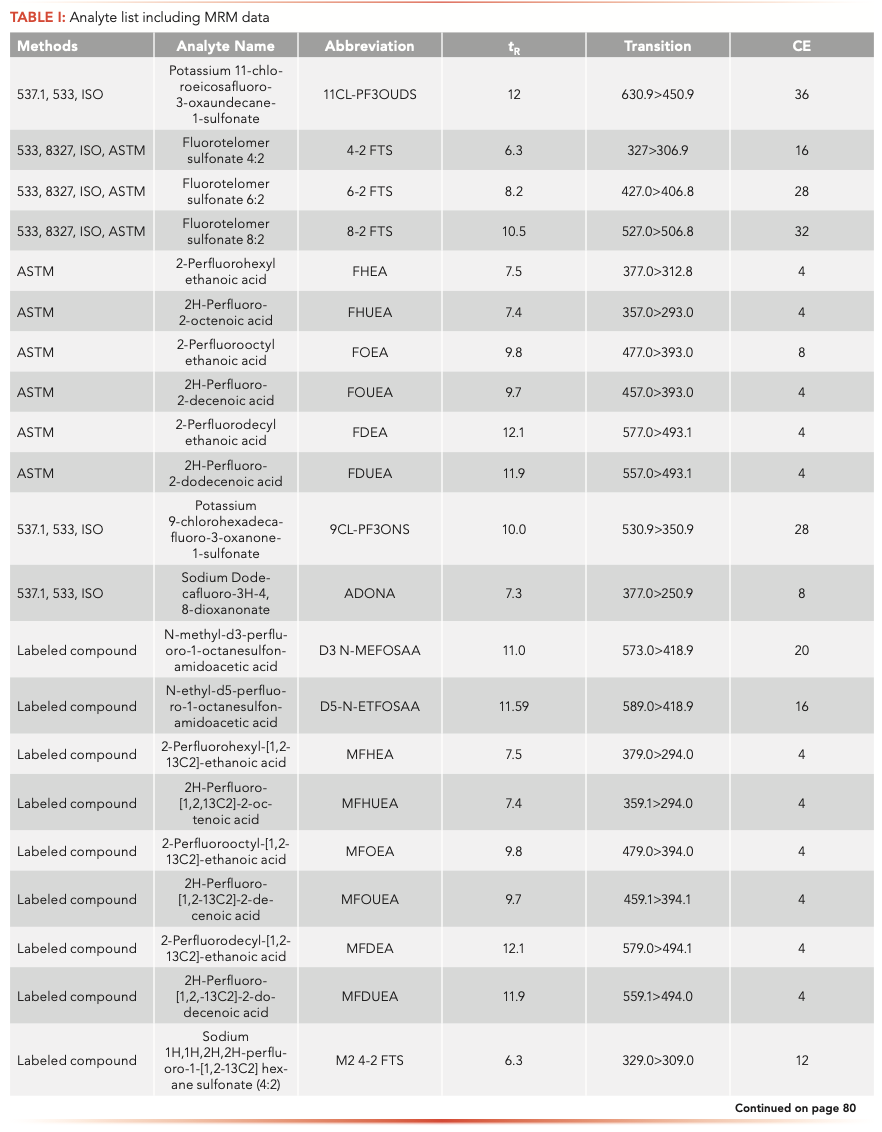
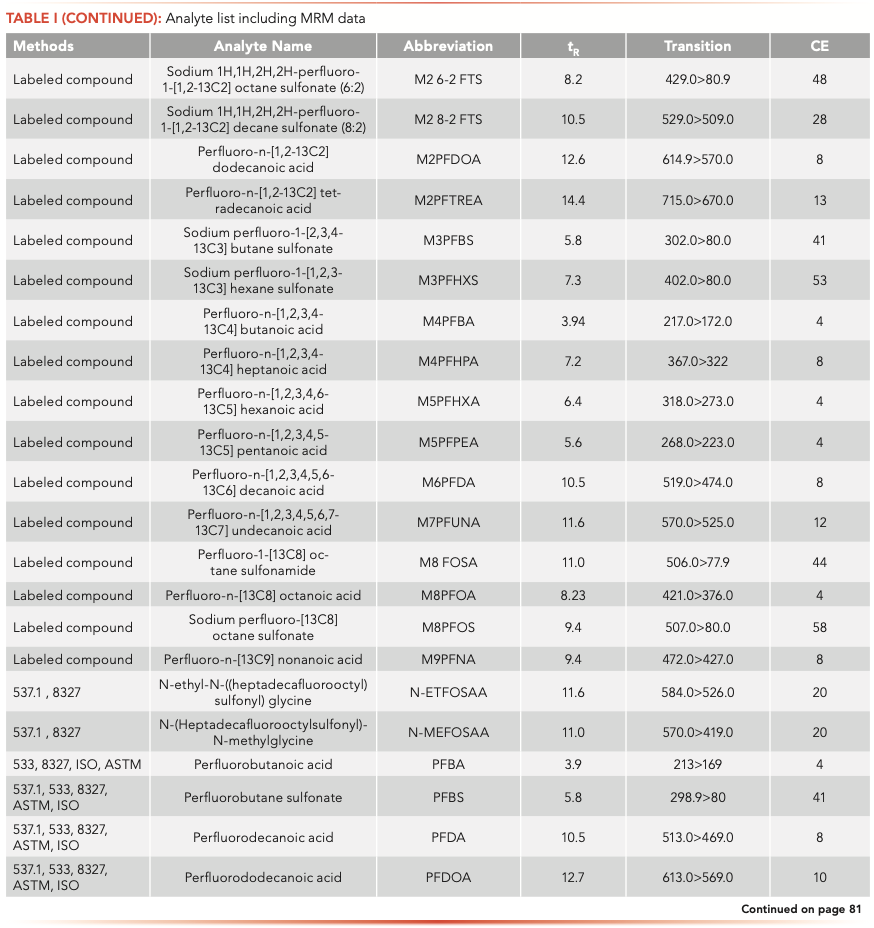
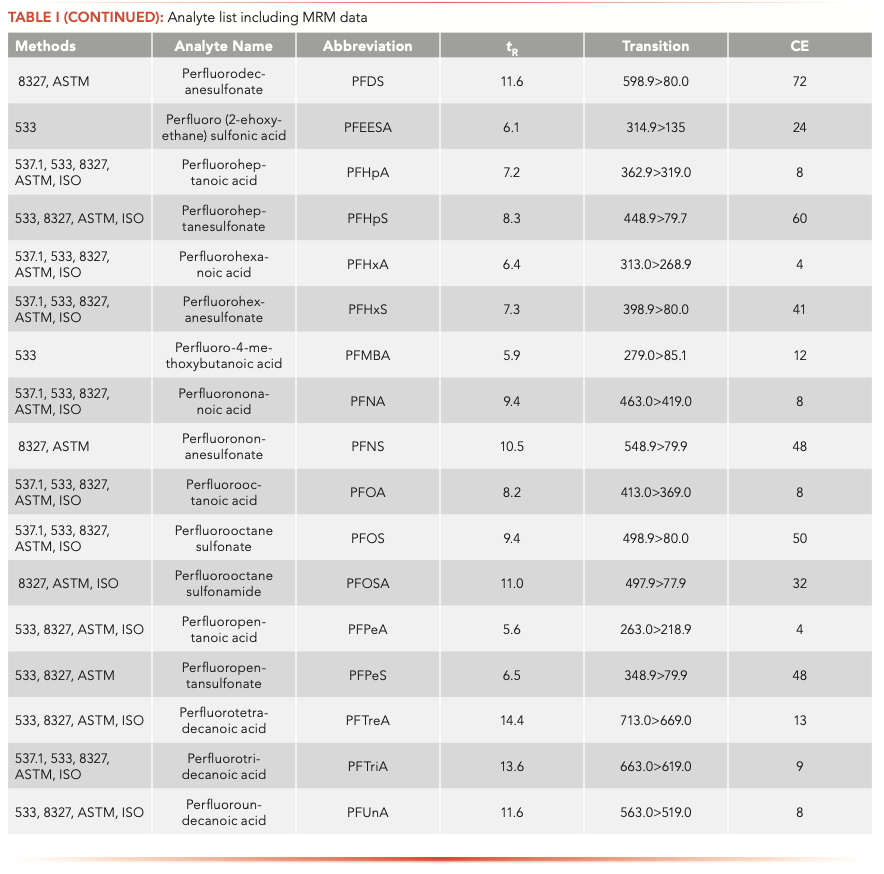
Native and labeled PFAS compounds (Table I) were acquired from Wellington Laboratories (Guelph, Ontario, Canada). Calibration and fortification experiments were prepared with 18 MΩ water. Surface water, drinking water, and wastewater effluent samples were all collected in Wilmington, Delaware.






Sample Preparation
First, 5 mL samples were collected in polypropylene centrifuge tubes. Labeled surrogates were added and the samples were diluted with 5 mL volume of methanol and inverted for 2 min. The entire sample was then filtered through a 0.2 μm syringe filter followed by acidification with 10 μL of acetic acid. 1 mL of the sample was then placed into a polypropylene vial and closed with polypropylene caps, now ready for LC–MS analysis. Both polypropylene and deactivated glass vials were tested, but longer chain compounds showed significant sorption to glass vials with time in the autosampler.
Instrumental Analysis
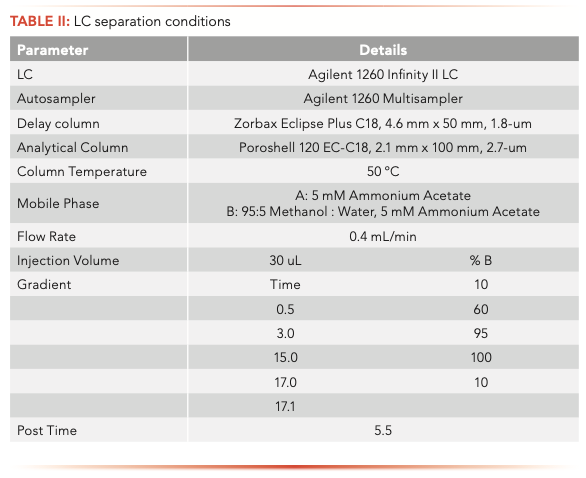
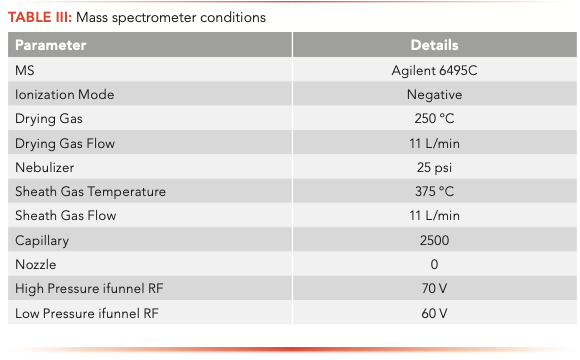
The samples were analyzed on an Agilent 1260 Infinity II liquid chromatograph (LC) equipped with a high-speed binary pump and multicolumn compartment coupled to an Agilent 6495C Tandem Quadrupole LC–MS. For this experiment, 57 PFAS (35 native and 25 internal standards) were analyzed simultaneously using dynamic multiple reaction monitoring (DMRM) with data acquisition and processing done using MassHunter software. All compound parameters and MS source parameters were optimized automatically using the Agilent Compound Optimizer and its source optimizer features. LC and MS parameters are provided in Tables II and III, respectively.




Quantification
Isotopic analogues were associated with native compounds. If a labeled compound was not available, compounds were corrected by either PFOA or PFOS, depending on whether the target was a sulfonate or an acid (that is, PFNS was corrected M8PFOS).
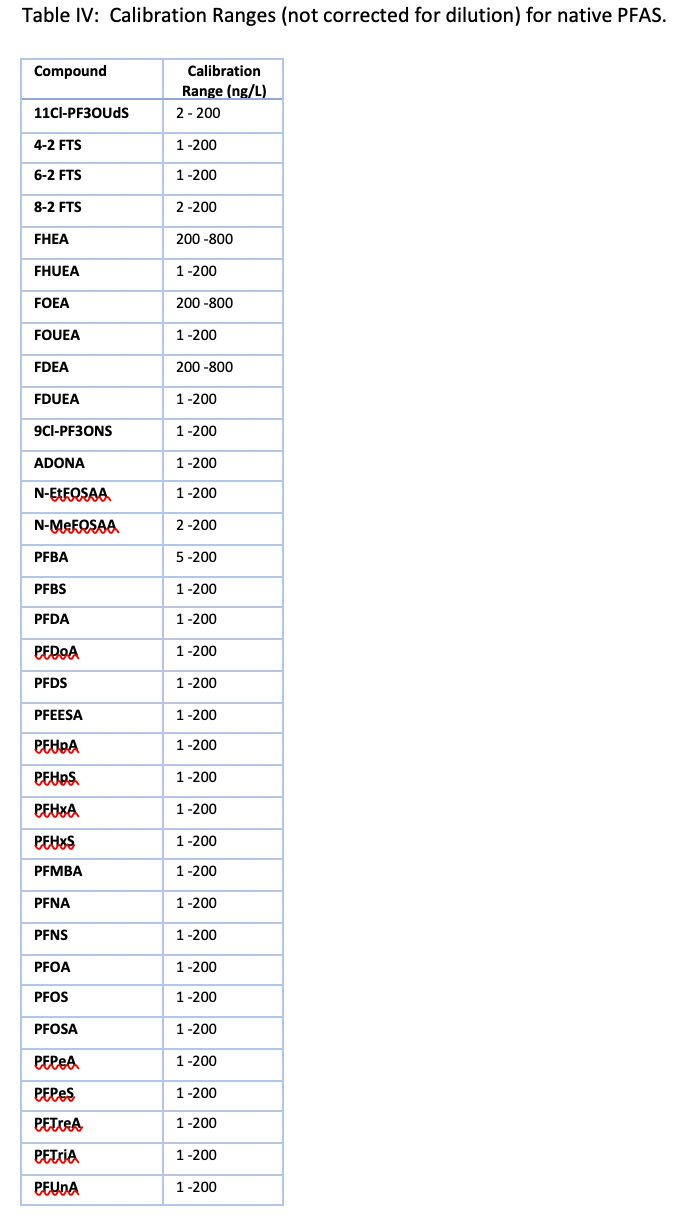
All calibration curves were linear with the origin ignored and 1/x weighting used with a minimum of seven points. Ranges are shown in Table IV and for most PFAS the lower limit of quantification of 2 ng/L was chosen. The fluorotelomers had a higher calibration range of 200–8000 ng/L because of lower sensitivity, but this is consistent with the EPA 8327 and ASTM 7979 methods.


Method Performance Evaluation
Method Detection Limits (MDL)
Over the course of three different days, a total of nine reagent water samples were spiked with native PFAS (see Table IV) concurrently with nine method blanks. Samples were prepared as described in the sample preparation section. MDLs were calculated as described in equation 1:
MDL = Standard deviation of replicates × Students t-valuen-1 [1]
Matrix Recovery
To investigate the effect of matrix on method performance and viability of the method to different waters, three different water types were tested. Within each group, blanks and spikes were processed. Surface water (n = 3), wastewater effluent (n = 4), and reagent water (n = 3) were prepared as described in the sample preparation section and analyzed with the method. For compounds with labeled surrogates, method performance was evaluated by percent recovery of isotopic analogue. Compounds that did not have a labeled analogue percent recovery were calculated from spike recovery with consideration of whether the compound was present in the blank of that water type.
Instrument Robustness
Wastewater effluent is the dirtiest matrix in the study and was used to determine instrument robustness. The wastewater effluent samples were injected continually for 36 h, bracketed every 10 samples by a continuing calibration check (CC) at 80 ng/L. The raw and uncorrected response of the CC was compared across the course of the worklist to evaluated how continual introduction of matrix affected instrument response and performance of the MS. Method performance specification require deviations to remain less than 30%.
Results
Background and Compound Separation
Instrument background was not noticed for all the PFAS after the installation of the PFAS elimination kit and delay column. The 35 native PFAS analyzed in this method were separated using an Agilent Poroshell 120 EC-C18 column (with column dimensions of 2.1 mm x 100 mm, 2.7-μm). Figure 1 shows the individual MRMs of the PFAS analyzed at a 2 ng/L concentration standard. It can be noticed that for most compounds lower concentrations can be analyzed. PFBA does show peak fronting, likely because of a large volume injection of a solvent other than the initial gradient conditions (nominally 5% methanol). Different mobile phase combinations and columns were tried; however, 5 mM ammonium acetate provided the best sensitivity for the widest range of compounds. One study showed good PFBA peak shape with a Zorbax SB-C18 column; however, the mobile phase pH was lowered with acetic acid to maintain the improved peak shape (18). With the additional analytes added and the goal of ultimate sensitivity, it was decided to compromise PFBA peak shape for enhanced sensitivity.
Method Detection Limits
In the ASTM 7979 and EPA draft 8327 method, the lowest reporting limit is 10 ng/L for some PFAS, while many are higher (17). The fluorotelomer acids (FTAs) have a lowest limit of quantification in the method at 200 ng/L. In this method, employing the 6495C tandem quadrupole LC–MS, calculated method detection levels were below 10 ng/L for all PFAS tested except the three fluorotelomer acids. In fact, greater than 80% of the PFAS tested had method detection levels less than 2.5 ng/L, which is four times lower than the 10 ng/L level suggested for any PFAS in the ASTM or EPA methods. This shows the enhanced sensitivity of the instrument. The benefits of sensitivity do not extend only to lower reporting levels which may be critical in future proofing a laboratory against expected reduction in regulatory PFAS limits. Additionally, the enhanced sensitivity allows a laboratory to reduce injection volumes or extract less sample that can lead to several benefits, like less contamination going into the MS instrument and lower frequency of maintenance along with ancillary benefits of lower sampling and shipment costs required because of the smaller sample usage.
Matrix Spike Recovery
Figure 2a shows the surrogate recovery of the 25 isotopically labeled PFAS spiked at 80 ng/L in the three different water matrices tested. The average recovery and relative standard deviation (n = 4) of the samples is plotted in the figure. All water types, namely reagent water, surface water, and wastewater, were within the ASTM and USEPA method guidelines of 100% ± 30% surrogate recovery. Average recovery of the surrogates was much closer to a 100% for most analytes with ranges of 87–104% in reagent water, 83–107% in surface water, and 83–107% in wastewater.
Figure 2b shows the average recovery of nine additional PFAS tested that did not have a matching isotopically labeled standard in the three water matrices. Perfluoro-4-methoxybutanoic acid (PFMBA) was present at low levels in the unspiked wastewater replicates so recoveries were corrected to take into account the background in the wastewater sample. All other compounds had clean matrix blanks with concentrations lower than limits of detection and recoveries were within method guidelines. All nine had recoveries without any correction between 70–130% in all three water matrices, indicating good recovery and very accurate detection.
Instrument Robustness
Because this method uses minimal sample preparation and cleanup, it is critical that the instrument be robust and stable across long periods so there is no drift or drop in sensitivity. Additionally, it is critical for the instrument to keep a stable response and not let matrix effect the MS instrument, which will lead to frequent cleaning, maintenance, and downtime. The ASTM 7979 method also uses a similar sample preparation but does not require use of internal standards so true and raw recovery is critical for the MS instrument to maintain across a large run. In this study, a worklist of wastewater effluent samples were run across 36 h and after every 10 samples a continuing calibration standard of all 35 PFAS was run at 80 ng/L. The raw and uncorrected responses of each analyte are normalized and plotted across the 36 h in Figure 3. The system showed excellent robustness across repeated injections of wastewater. Of the 35 analytes, 61% had RSDs that remained below 6% for the duration of the 36 h test and 85% of the analytes has RSDs that remained below 10% without any correction of response with internal standards (ISTDs). Only two compounds exceeded the method guidelines of 30%, 2H-Perfluoro-2-dodecenoic acid (FDUEA) and 2-perfluorodecylethanoic acid (FDEA). Those two compounds, as well as some of the longer chain PFCAs (PFTreA, for example), showed a downward trend after 24 h had passed. Rather than instrument performance, an alternative explanation for this trend is that these compounds are less soluble and stable in the 50:50 methanol:water solution and could be falling out of solution. Re-vortexing the autosampler vials might resolubilize them and could be tested as one potential solution. The multisampler was kept at room temperature during analysis in an attempt to maintain solubility. The results, however, indicate excellent robustness of the system under long-term stress of heavy matrix wastewater samples.
Conclusions
With the regulatory landscape around PFAS changing globally and driving expected measurement levels down, in addition to a significant increase in sites with potential PFAS contamination coming to light, it is critical to have analytical methods that offer ultimate sensitivity, while maintaining robust and reliable quantification data even when challenging matrices are tested and minimal sample preparation is employed to have high-throughput. The method demonstrated here analyzes more than 50 PFAS in different and challenging water matrices, achieving low detection levels with high data quality and robustness, meeting and exceeding established guidelines by standard methods.
References
(1) Z. Wang, J.C. DeWitt, C.P. Higgins, and I.T. Cousins, Environ. Sci. Technol. 51(5), 2508–2518 (2017).
(2) R.C. Buck, J. Franklin, U. Berger, J.M. Conder, I.T. Cousins, P. de Voogt, A.A. Jensen, K. Kannan, S.A. Mabury, and S.P.J. van Leeuwen, Integr. Environ. Asses. 7(4), 513–541 (2011).
(3) E. Kissa, J. Fluor. Chem. 66(1), 5–6 (1994).
(4) J.M. Conder, R.A. Hoke, W. De Wolf, M.H. Russell, and R.C. Buck, Environ. Sci. Technol. 42(4), 995–1003 (2008).
(5) N. Tominaga, S. Kohra, T. Iguchi, and K. Arizono, J. Health Sci. 50(5), 545–550 (2004).
(6) V.P. Beškoski, K. Yamamoto, A. Yama- moto, H. Okamura, M. Hayashi, T. Nakano, C. Matsumura, K. Fukushi, S. Wada, and H. Inui, Chemosphere 170, 260–265 (2016).
(7) M.H. Cai, Z. Zhao, Z.G. Yin, L. Ahrens, P. Huang, M.G. Cai, H.Z. Yang, J.F. He, R. Sturm, R. Ebinghaus, and Z.Y. Xie, Environ. Sci. Technol. 46(2), 661–668 (2012).
(8) T.L. Coggan, D. Moodie, A. Kolobaric, D. Szabo, J. Shimeta, N.D. Crosbie, E. Lee, M. Fernandes, and B.O. Clarke, Heliyon 5(8), e02316 (2019).
(9) L.A. Schaider, S.A. Balan, A. Blum, D.Q. Andrews, M.J. Strynar, M.E. Dickinson, D.M. Lunderberg, J.R. Lang, and G.F. Peaslee, Environ. Sci. Technol. Letters 4(3), 105–111 (2017).
(10) Q. Jiang, R.M. Lust, M.J. Strynar, S. Dagnino, and J.C. DeWitt, Toxicology 293(1–3), 97–106 (2012).
(11) Risk Management for Per- and Polyfluoroalkyl Substances (PFAS) under TSCA. https://www.epa.gov/assessing-and-managing-chemicals-under-tsca/risk-management-and-polyfluoroalkyl-substances-pfas 2006.
(12) M. Strynar, S. Dagnino, R. McMahen, S. Liang, A. Lindstrom, E. Andersen, L. McMillan, M. Thurman, I. Ferrer, and C. Ball, Environ. Sci. Technol. 49(19), 11622–11630 (2015).
(13) T.L. Coggan, T. Anumol, J. Pyke, J. Shimeta, and B.O. Clarke, Anal. Bioanal. Chem. 411(16), 3507–3520 (2019).
(14) USEPA 537.1: Determination of Selected Per- and Polyfluorinated Alkyl Substances in Drinking Water by Solid Phase Extraction and Liquid Chromatography/ Tandem Mass Spectrometry (LC–MS/MS). https://cfpub.epa.gov/ si/si_public_record_Report.cfm?dirEntryId=343042&Lab=NERL 2018.
(15) USEPA 533: Determination of Per- and Polyfluoroalkyl Substances in Drinking Water by Isotope Dilution Anion Exchange Solid Phase Extraction and Liquid Chromatography/Tandem Mass Spectrometry. https://www.epa.gov/dwanalyti- calmethods/method-533-determination-and-polyfluoroalkyl-substances-drinking-water-isotope 2019.
(16) Validated Test Method 8327: Per- and Polyfluoroalkyl Substances (PFAS) Using External Standard Calibration and Multiple Reaction Monitoring (MRM) Liquid Chromatography/Tandem Mass Spectrometry (LC/MS/MS). https://www.epa.gov/hw-sw846/validated-test-method-8327-and-poly-fluoroalkyl-substances-pfas-using- external-standard 2019.
(17) ASTM D7979 - 19 Standard Test Method for Determination of Per- and Polyfluoroalkyl Substances in Water, Sludge, Influent, Effluent, and Wastewater by Liquid Chromatography Tandem Mass Spectrometry (LC/ MS/MS). https://www.astm.org/Standards/D7979.htm
(18) J. Zweigenbaum and H. Zhao, Simplified and Fast Analysis of Per-and Polyfluoroalkyl Substances in Non-potable Waters. Agilent Application Note, 5994-0678EN 2019.
Emily Parry is a LC–MS Applications Scientist with Agilent Technologies, Inc. Tarun Anumol is the Director of Global Environment and Food Applied Markets at Agilent Technologies, Inc. Direct correspondence to: emily.parry@agilent.com














HILIC Peptide Retention Times Predicted Using New Approach
October 29th 2024Manitoba Centre for Proteomics and Systems Biology scientists produced a new means of predicting peptide retention times for hydrophilic interaction liquid chromatography (HILIC) at acidic pH in formic-acid based eluents.

")




