Asymmetric Flow Field-Flow Fractionation (AF4) with Multi-Angle Light Scattering (MALS) for High-Throughput Protein Refolding
LCGC Europe
One of the biggest problems facing researchers involved in pharmacogenomics is analysing the recombinant proteins of interest to monitor if they are in a folded state. This article describes a rapid and economical method using asymmetric flow field-flow fractionation combined with multi-angle light scattering (AF4–MALS) to characterize refolded proteins, which overcomes some of the disadvantages associated with other techniques.
Since completion of the Human Genome Project, where approximately 100000 human proteins were identified, a large number have been highlighted as potential targets for pharmaceutical and biotechnology companies. These companies require the use of high-quality proteins for tools in assay development, for protein structural studies and to identify biological functions. However, of these targets, less than 500 are actually commercially available. The problem facing researchers is how to get sufficient quantities of a large number of proteins both quickly and at a reasonable cost.



This need for the large-scale production of recombinant proteins requires that the proteins be in the native and active conformation (thus in the folded state). Since the genes' and corresponding proteins' biological functions are unknown, it is not possible to determine whether it is in a native or non-native state via bioassay.
Alternative methods for monitoring protein refolding, for example, reverse phase high performance liquid chromatography (RP–HPLC), size-exclusion chromatography (SEC) and fluorimetry are subject to a range of limitations. This article will review the use of asymmetrical flow field-flow fractionation (AF4) combined with multi-angle light scattering (MALS) by the Protein Structure Unit, Discovery Technologies, Novartis Institutes for Biomedical Research, CH-4002, Basel, Switzerland, as a viable method for characterizing refolded proteins.
High-Throughput Protein Refolding
Escherichia coli (E.coli) remains the expression of choice for structural studies. This is because of the homogeneity of proteins produced, expression levels, the speed of manufacture and ease of labelling. However, for a given number of targets, a certain percentage will be soluble and some will be insoluble (between 40–60%) thus are expressed as insoluble materials known as inclusion bodies (IBs). It is easy to isolate, extract and purify these IBs via a process called solubilization, the problem lies, however, with extracting the functional, correctly folded proteins by high-throughput refolding.
Refolding is a process that results in a change in protein conformation from an unfolded to folded (native) state. In order to achieve this, initially the IBs are solubilized by agents such as detergents, chaotrophs or urea. Solubilized IBs contain impurities, which may interact with the expressed protein and thus interfere with its refolding. In addition the high concentration level of the denaturant that was used to solubilize the IB causes the proteins to be disordered (unfolded), solvated and flexible. Thus, it is necessary to reduce the denaturant concentration and change the solution conditions in a way to aid the refolding process. The process to find the best protocol is based on trial and error experiments, in which a set of chemicals and conditions are tested.
Refolding Read-Out Methods
A number of variables can be included to maximize the chances of success in the refolding process, for example, buffer additives, different pHs and artificial chaperones. Once refolding has occurred however, the difficulty lies with determining whether the refolding has been successful, for example, distinguishing an optically clear solution containing folded protein from a clear solution containing denatured or aggregated proteins.
Techniques such as RP–HPLC and turbitometry are unlikely to distinguish the two; circular dichroism spectroscopy (CD) is insensitive and provides low throughput. Fluorimetry, however, can differentiate between folded and unfolded proteins but it needs higher protein concentrations that are not compatible with a 96-well plate format. Additionally, sensitivity depends heavily on the content of tyrosine and tryptophan residues in the protein, as well as on quenching effects caused by some of the buffer additives used. Therefore, the fluorimetry method does not work consistently well for all types of proteins and protocols, depending on amino acid composition and additives used. It is important to note that fluorimetry is not an end-point read-out, but can also be used to compare samples.
Size-exclusion chromatography (SEC) cannot tolerate the high amounts of aggregates that have to be injected on the column. Some of the additives used are aggressive and can damage column packings. Column plugging leads to pressure increase, reproducibility problems and the lifetime of the column is greatly decreased.
Flow Field-Flow Fractionation
What, then, is the solution? Novartis used an Eclipse™ Field-Flow Fractionation system and a miniDAWN multi angle light scattering detector (both from Wyatt Technology, Santa Barbara, USA), with an Agilent HP1100 to provide flow, degasser, UV detector and autosampling capabilities.
The Eclipse works on the basis of flow field-flow fractionation, but the technical implementation makes the use of the system as straightforward as SEC or HPLC.
Macromolecular characterization via AF4 is a non-destructive, one-phase separation mechanism to determine molecular and macromolecular weights and particles in solution. As with classic chromatography, sample components elute at given retention times which are related to a range of physiochemical properties of the retained species. However, in contrast to classic chromatography, an AF4 separation works only on hydrodynamic properties and does not involve interaction with a stationary phase.
Separation is achieved by the differential flow of a solvent in a channel, consisting of two plates that are separated by a spacer foil; the plates are bolted together. The upper channel plate is impermeable, whilst the bottom plate is permeable and made of a porous frit material. An ultrafiltration membrane with a typical size barrier of 10 kD, covers the bottom plate to prevent the sample from penetrating the channel.
In the first step the sample is introduced into a solvent stream which is injected into the channel. As a necessary part of the separation procedure, the sample is first focused in the channel region near the injection port. This is achieved by splitting the channel flow into two components, each of which enters the channel from opposite ends. The flows are adjusted so that they meet close to the injection port and at this point the flow direction is perpendicular towards the porous bottom of the channel. The sample is driven towards the bottom wall and concentrated close to the membrane. Diffusion associated with Brownian motion creates a counteracting motion. Within a couple of minutes a stationary equilibrium is established, in which the two forces balance out for each particle size at a different distance from the bottom wall. Smaller particles, which have higher diffusion rates and are subject to lower friction force, reach an equilibrium position further away from the bottom wall. Bigger particles or molecules will be driven closer to the bottom wall.
In the second step involving elution, the flow pattern is restored to go from inlet to channel outlet and transport the sample along the channel. The flow is laminar, which means it has a parabolic velocity profile with transport speed higher away from the bottom wall. This is like in a river where boats will float downstream much faster in the middle of the stream. The sample components are like boats in a floating race; smaller particles, farther away from the "shore", will travel much faster and elute first, before the larger ones. This is exactly the opposite of a size exclusion (SEC) or gel permeation (GPC) separation in which the large molecules elute first.
The process is rapid and gentle because of low shear forces and the absence of any stationary phase. In comparison to SEC, AF4 displays higher selectivity and has a much broader application range.
Multi-Angle Light Scattering (MALS)
The MALS detectors used in this study are the only absolute multi-angle light-scattering detectors available. This means that they measure molar mass directly from the scattered light intensity without reference to a standard or "known" molar mass.
In the application of determining protein monomer molar mass and detection of aggregates, sensitivity of the light scattering detector is critical. Proteins are present in very small concentrations and only a few microlitres can be used for the measurement. As scattering intensity is proportional to concentration times molar mass, the resulting light scattering peak will be orders of magnitude smaller for the monomer compared to aggregates, because the latter have high molar masses of millions of Daltons versus tens of thousand Daltons for the native protein. So the detection task is formidable and needs a high dynamic range of the light-scattering photometer. The AF4-MALS technology proved to stand up to this challenge and provided accurate data that could be used to identify the protein monomers and aggregates in one separation run. The unique flow cell structure combined with a sophisticated optical bench and read head proves to be critical in processing such small light-scattering peaks.
AF4–MALS
AF4-MALS combines high-resolution separation with absolute detection of molar masses and proved more than adequate to identify and quantify refolded monomeric proteins. Runs were typically completed within 12 min, producing results that were highly reproducible. Monomers and aggregates could be reliably separated. The system was automated using an HPLC autosampler and was sensitive enough to provide good signals with as little as 1–2 μg protein.
Using MALS, Novartis were able to obtain accurate mass calculations of the refolded protein monomers — typically within 10% of the nominal value. Yield quantification enabled a ranking of constituents of the folding matrix to asses the most productive refolding protocol for the specific protein. For example monomers and dimers can be detected and quantified as demonstrated in Figure 1.


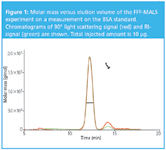
Figure 1: Molar mass versus elution volume of the FFF-MALS experiment on a measurement on the BSA standard. Chromatograms of 90° light scattering signal (red) and RI-signal (green) are shown. Total injected amount is 10 μg.
Results
To evaluate precision, Novartis performed multiple consecutive runs of a protein and verified that the monomer/dimer quantification was reproducible, In a model study, the AF4–MALS method was compared to HPLC quantification of refolded monomer. It was shown that whereas HPLC reported constant protein values, the AF4-MALS method could discriminate between refolding protocols and show valid variations in monomer recovery.
Conclusion
AF4 combined with MALS is a robust and reliable means of ranking refolding conditions. Although Novartis had tested several methods for determining whether or not proteins have been refolded, this method was by far the most reliable and informative characterization tool. AF4–MALS turned out to be applicable to a wide range of refolding protein projects and produced consistent results. Other methods provided misleading results in one or more instances; over the sample set tested, AF4 was 100% reliable. Additionally, AF4 was found to be an excellent method for characterizing proteins produced for structure elucidation.
By using AF4–MALS, Novartis has refolded several new proteins, as well as significantly improving several of its refolding protocols. Moreover, hits have noticeably varied from protein to protein, which means that only by running through the test matrix the optimal protocol could be found. Further developments in the folding screen look promising, and Novartis will implement continuous modification/optimization to the screen. It will also need to undergo some expansion.
Acknowledgements
Thanks to René Hemmig and Paul Ramage at Novartis Pharma AG for providing the material for this article.
Christoph Johann is Managing Director of Wyatt Technology Europe GmbH, located in Germany. He received his PhD in polymer science in 1985 and founded the Wyatt Technology Europe organization in 1993. He started working with FFF in 1992 and sold the first commercial FFF systems in Europe in the following years. Since 2003 Wyatt Technology has been developing and producing the Eclipse AF4 system in Germany.
Paul Ramage studied biochemistry at the University College of Wales, Aberystwyth, UK. After completing an MSc in Biotechnology at Birmingham University, UK, he went on to do a PhD in Biochemistry at Edinburgh University, UK. After 5 years as a senior protein chemist at Wellcome he moved to Switzerland in 1990, joining the then Sandoz AG, which several years later became Novartis following a merger with Ciba-Geigy. He is currently Head of the Protein Preparation Group in the Protein Structure Unit.
After training in the Sandoz Technician School as a synthetic chemist, Rene Hemmig joined Sandoz AG in 1980 where he worked in a number of process developement, analytical chemistry and biochemistry laboratories. After several years specializing in protein analysis in the then Biotechnology Development Group, he joined Paul Ramage's laboratory in 1990 where he has developed protein purification methods, in particular in protein refolding.


 with Multi-Angle Light Scattering (MALS) for High-Throughput Protein Refolding")














AOAC International Awarded NIST Grant for Developing Drug Testing Standards
October 31st 2024The grant will be part of a new collaborative scientific initiative to address the need for standards that define the desired performance of lateral flow immunoassay test strips to detect illicit drugs in tablets and powders.

AI and GenAI Applications to Help Optimize Purification and Yield of Antibodies From Plasma
October 31st 2024Deriving antibodies from plasma products involves several steps, typically starting from the collection of plasma and ending with the purification of the desired antibodies. These are: plasma collection; plasma pooling; fractionation; antibody purification; concentration and formulation; quality control; and packaging and storage. This process results in a purified antibody product that can be used for therapeutic purposes, diagnostic tests, or research. Each step is critical to ensure the safety, efficacy, and quality of the final product. Applications of AI/GenAI in many of these steps can significantly help in the optimization of purification and yield of the desired antibodies. Some specific use-cases are: selecting and optimizing plasma units for optimized plasma pooling; GenAI solution for enterprise search on internal knowledge portal; analysing and optimizing production batch profitability, inventory, yields; monitoring production batch key performance indicators for outlier identification; monitoring production equipment to predict maintenance events; and reducing quality control laboratory testing turnaround time.






