Analytical Tools for Characterization of Biotechnology Products and Processes
LCGC North America


In the first installment of LCGC's newest column, the authors address the topic of analytical biotechnology.
Biotech is still a very young science, industry, practice, and pursuit, and there is much yet to be discovered and commercialized. More and more biopharmaceutical (biopharma) companies are seeking regulatory approvals, more and more of pharmaceutical R&D are going into biopharma, and still more growth is expected in the coming years. We hope that this column can help foster this continued research, growth, and development in the future. Reader feedback will be essential for us to ensure that we address timely topics that would be of interest and value to this column's and magazine's readership.



Ira S. Krull
Column Scope
This column series will present where the biotech industry has been, where it stands today, and where it may be going tomorrow. The scope of coverage will include all aspects of the modern biopharmaceutical industry, but we will also discuss topics in other areas of biotechnology such as biofuels, biosensors, and nanobiotechnology. Numerous reliable sources of information already exist about the current status of biotechnology and the biotech industry, in the U.S. and abroad, and many states, regions, and countries have biotech councils or organizations trying to promote the industry and educate the public (1–6). There are also a very large number of texts and journals devoted to the different applications of biotechnology (7–11).



Anurag S. Rathore
We also hope to emphasize in future columns the ongoing efforts within the U.S. Food and Drug Administration (FDA) (European Medicines Agency [EMEA] already has established guidelines on the topic), and the US Pharmacopeia (USP) to establish regulatory requirements for the successful and safe introduction of biosimilars (as they are designated in Europe) and follow-on biologics (FOB) (as designated in the U.S.), and how these can differ in such requirements from the original, proprietary drug products. We can readily envisage some lively discussions and debates about what should or must go into these newer regulatory requirements for biotech, lookalike, FOB products. As the U.S. Congress and FDA now wrestle with how to regulate such FOBs, they eventually must legislate to the industry what will be expected in newer submissions of FOBs and why, or legal wrestling between the innnovator company and the FOB will just delay the introduction and perhaps acceptance of FOBs more than necessary. Such legislation and guidelines are needed to avoid this unwanted scenario. How can this be brought about successfully and more quickly could well be the subject of an upcoming column.
Background Information
Ira S. Krull is Professor Emeritus of Chemistry and Chemical Biology at Northeastern University (Boston, Massachusetts) and has been on LCGC's Editorial Advisory Board for more than 10 years. He has been a coeditor of "Validation Viewpoint," another LCGC column, since its inception. He has taught at the undergraduate and graduate levels for more than 30 years, mainly related to analytical chemistry, has had an active R&D group of students, postdocs, and visiting professors, has published over 300 scientific or technical articles in refereed journals, magazines, and textbooks, and has presented over 500 papers at meetings around the world. He also has coedited or coauthored several textbooks in various areas of analytical R&D. In addition, he has given numerous training courses at scientific meetings and to private industry, and has consulted for many pharma and biopharma firms, around the world. His interest in analytical biotechnology and biopharmaceutical regulatory science remains active to this day. He has a Ph.D. in organic chemistry from New York University, New York.
Anurag S. Rathore is a consultant of Biotech CMC Issues. He is also a faculty member with the Department of Chemical Engineering, Indian Institute of Technology (Delhi, India). His previous roles included leadership roles at Amgen, Inc. (Thousand Oaks, California) and Pharmacia Corporation (St. Louis, Missouri). His areas of interest include process development, scale-up, technology transfer, process validation, process analytical technology, and quality by design (QbD). He has authored more than 100 publications and presentations in these areas and serves on the editorial advisory boards for PDA Journal of Science and Technology, Biotechnology Progress, Biopharm International, Pharmaceutical Technology Europe, and Separation and Purification Reviews. Dr. Rathore has edited books such as Quality by Design for Biopharmaceuticals: Perspectives and Case Studies (2009), Elements of Biopharmaceutical Production (2007), Process Validation (2005), Electrokinetic Phenomena (2004), and Scale-up and Optimization in Preparative Chromatography (2003). He has a Ph.D. in chemical engineering from Yale University, New Haven, Connecticut.
While we have somewhat different, formal education pedigrees and experiences, we have both been actively involved in biotechnology for quite some time. While one of us (Dr. Krull) has been involved more directly in academic R&D, the other has been more involved in the biotech industry R&D.
Analytical Biotechnology
In the first column of this series, we address the topic of analytical biotechnology. This includes (but is not limited to) determination of a protein's purity, primary–secondary–tertiary structures, amino acid sequence, amino acid analysis, molecular weight determinations, aggregation, posttranslational modifications (PTMs) and their characterizations, profiling of glycan modifications, biological activity, immunogenicity of drug substances (all related to the drug substance characterization), impurity profiling, stability studies, formulation development and testing, and biological activity or efficacy. We intend to discuss the very latest advances in analytical instrumentation for improved characterizations of biotechnology products, and not just proteins, but also carbohydrates, nucleic acids (DNA and RNA), and related materials. A major goal is to provide up-to-date descriptions and discussions of modern analytical instrumentation and methodologies that are of growing importance within the biotechnology industries around the world. This will include, in this and future columns, all relevant separations technologies, for example, ultrahigh-pressure liquid chromatography (UHPLC), field-flow fractionation (FFF), supercritical fluid chromatography (SFC), gas chromatography (GC), one-dimensional gel electrophoresis (1DGE), two-dimensional gel electrophoresis (2DGE), multidimensional liquid chromatography (MDLC), liquid chromatography–mass spectrometry (LC–MS), and LC–MS-MS.
Fundamental Differences from Small Molecule Pharmaceuticals
Size and complexity: Most biotech products today are usually not single entities, as is the case with insulin or small peptides. They are often complex mixtures of proteins, fusion proteins, supramacromolecular complexes, and others. It is not just that such mixtures must be defined, resolved, and each component structurally characterized as best possible, but that there are often an unknown number of variants or isoforms that make-up, collectively, the total drug product.
The requirements for partial or complete characterization of biotech drug products has grown by leaps and bounds over small molecule analytics, to the point where regulatory agencies recognize the differences (28). Well-characterized biopharmaceutical products (WCBP) has become a quite common analytical phrase in the biotech industry today, as well as within most regulatory agencies (14). Figure 1 indicates the usually accepted, biopharmaceutical product development cycle, from having a target identified in the discovery phase all the way to the postapproval stage and comparability. QbD is intended to be utilized throughout the product development cycle, as possible (29,30). As indicated, having better and faster analyses will reduce a drug's development timelines and permit for more rapid process improvements. Whenever analytical methods can be done faster and with higher sample throughput, this leads to a more rapid, overall development of the drug's timelines. All of these things will lead to reduced development costs and, it is hoped, improved profits and stock pricings.


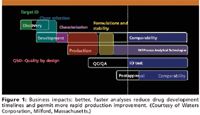
Figure 1
Expression of Protein Products
In the modern biotech world, chemical protein synthesis is not viable, though small peptides are still produced by solid phase synthesis much of the time and often with high purity and yields. In the absence of an analogous polymerase chain reaction (PCR) method for duplicating proteins, protein production is accomplished today with recombinant deoxyribonucleic acid (DNA) expression systems. Plasmid insertions into specific DNA strands and then insertion of these into an animal, tissue culture, or animal–plant cell, is now beyond commonplace. And bacterial, animal, and plant cells are being used almost everywhere today, to express any variety and number of proteins, peptides, antibodies, fusion proteins, and other biotech products (2–6). Plants are being used for extraction of their glycoproteins directly as drug substances, after suitable processing (6). It is questionable if this is true recombinant biotechnology or isolation of natural products. Other plant cells are being used to recombinantly express different, desired glycoprotein mixtures as drug substances, which can then be processed as drug products (23). Transgenic animal milk is now being used to produce recombinant products, in high purity and with economical efficiency of scale (37,38).
Orthogonal approach toward analysis: Just as the size, complexity, and number of variants has increased in going from small to larger molecules, so too, have the type and number of analytical tools and instrumentation. We no longer can rely just on UV, fluorescence, nuclear magnetic resonance (NMR), or MS, but often require hyphenated techniques, such as UHPLC–electrospray ionization (ESI)-MS-MS, high resolution MS (HRMS), high resolution NMR, FT-IR, size exclusion chromatography–multiangle laser light scattering (SEC–MALS), analytical ultracentrifugation, 2DGE (not as common as 1DGE in biopharma), MDLC, and others. Data handling requires sophisticated computer hardware and software, methods to interpret the protein's primary structure from its MS-MS sequencing routines, methods to define where a glycan resides on a protein's backbone, and what is the structure of that glycan. HRMS using MS instrumentation that did not exist 20 years ago is commonplace throughout the biotech world (35,36). It is now used, for example, to determine the nature of a particular variant glycoprotein, changes in location and nature of glycosylation patterns and changes in an amino acid in the protein backbone as a variant or isoform or PTM (31). Protein macromolecular complexes, as opposed to simpler protein aggregates (noncovalent associates), are now being assayed using ion mobility spectrometry (IMS) interfaced with time-of-flight MS (TOFMS) (IMS–TOFMS) to resolve mixtures of such complexes and to then determine intact MW and the components present in such complex complexes. Such macromolecular complexes might not pertain to biopharmaceuticals today, but they are possible drug targets.
The days of the single-quadrupole GC–MS instrument have given way to modern UHPLC–ESI-MS-MS (HRMS) tools for biotechnology products or capillary electrophoresis (CE)–ESI-MS-MS (39). Analytical instrumentation for the biotech industry will continue to get more and more complex, as in the latest series of orbitrap-related mass spectrometers. Who would have believed 20 years ago that which such instruments are capable of doing today for protein and PTM characterizations? The current suite of tools is indeed impressive! Separations have become faster, more efficient, better resolving, more reproducible and rugged than ever, with higher and higher peak capacities. We have available UHPLC, CE, FFF, 2DGE, MDLC, and others for resolving complex mixtures of recombinantly or naturally derived proteins, peptides, and antibodies. And, while it is not always possible or necessary to resolve all components of a desired mixture having certain biological properties, it is expected that we know their safety, efficacy, and clinical properties and can make them similar (qualitatively and quantitatively) with every batch and demonstrate batch-to-batch chemical equivalencies. Often, the process has come to define the product. We also have numerous sensitive and selective detection techniques (for example, FTMS, MALS) at the end of the separations scheme to give us specific, sensitive information about the nature and chemical structures of the different sample components. We can even do MDLC, fully automated, with online fraction collection, and FTMS-MS, at times along with peptide mapping and peptide mass fingerprinting, to tell us what variants of the parent protein were originally injected.
Where do the analyses occur?: Product and process characterization, both heavily dependent upon analytical methods, are two of the critical components that are required for commercialization of biotech products. Product characterization reveals the biochemical and biophysical nature of the product, as well as the nature of product-related substances and impurities. Thorough product characterization is a necessary precursor to determination of critical quality attributes (CQA), and the associated analytical methods that, in turn, can be utilized as in-process controls, specifications, and for stability and comparability testing (28). Process characterization focuses on understanding and defining the operating and design spaces for the process to achieve product with consistent CQA (29,30).
The development and commercialization of protein therapeutics involves migration of the product of interest through the various phases of product development (Figure 1). Following are some of the key activities related to analytical method development (32,33):
- Preclinical phase: Use of platform methods; testing focused on overall purity and product identification.
- Phases I/II: Method development and optimization; establishment of analytical schemes for product and the product intermediates; testing to include product, product related impurities, process related impurities, host cell related impurities and process reagents and additives; characterization of product and major impurities; stability studies.
- Phase III: Method qualification; finalization of overall testing and control scheme; characterization of product and other major and minor impurities; stability studies.
- Before initiation of process validation campaign: Method validation; support of process characterization studies; completion of the final product characterization package; stability studies.
- Before regulatory filing for final approval: Support of process validation campaign; execution of product comparability studies; stability studies.
- Postapproval: Support of lifecycle, management activities of the product including process monitoring; fulfillment of postapproval commitments; support of any postapproval process changes; upgrading of analytical technology leading to improvements in technology and practices.
Regulatory Differences and Challenges
In a future column, we wish to compare regulatory guidance from three of the major, biopharma regulatory bodies; U.S. FDA, EMEA, and JPMDA (Japanese Pharmaceuticals and Medical Devices Agency). Suffice it to say here that each major biopharma regulatory agency has set its own requirements and standards, often following the International Conference on Harmonization (ICH) guidelines and/or that of the USP. However, there are very real differences, at times, between these regulatory agencies, and for those that are seeking regulatory approvals from multiple jurisdictions, it is extremely important to understand the nuances in regulatory expectations. Figure 2 illustrates a task tree for the full structural characterization of a therapeutic protein (product). However, it is often not quite possible to obtain the "full" structural characterization mixtures of glycoproteins.


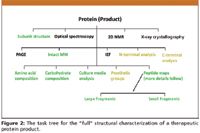
Figure 2
Challenges Faced During Analysis of Biopharmaceutical Products
One could argue that the overall success of the biotech industry has depended, to a very large extent, upon being able to do partial or complete characterization of the drug substance (DS) and the drug product (DP). The regulatory agencies expect and demand to see suitable product characterization, at first, and then accepted analytical methods for lot release testing and a demonstration of product comparability (15,16). Without these being thorough and complete, within the chemical manufacturing control (CMC), there is little chance of any new biotech products reaching the marketplace, even with successful clinical testing. Thus, long before clinical testing commences, a biotech firm must complete an appropriate level of analytical testing, at least for chemical characterization of the drug substances. The nature and extent of such testing is a function of time and evolution in the drug's progress to market, as illustrated in Figure 1.


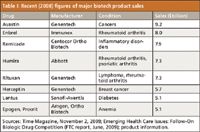
Table I: Recent (2008) figures of major biotech product sales
In the small drug molecule world, complete chemical and structural characterization of a new drug substance is relatively trivial and often is done successfully in a short period of time, at reasonable costs. This is never the case with complex mixtures of glycoproteins, which is what many, if not most, newer biotech-derived products have become (1,6) (Table I). There are any number of successful or about-to-be successful drug products on the market that are antibody derived, and though their glycosylation patterns might be simpler than others, they are still much more difficult to characterize fully than aspirin or insulin. Hence, in many cases, complete chemical and structural characterization of drug substances, just cannot be realized today (17,34). Figure 3 illustrates three LC–ESI-MS chromatograms for different glycopeptides derived from a single glycoprotein or a mixture of glycoproteins. Indicated are extracted, selected ion monitoring (SIM) mass chromatograms of the triply charged glycopeptides. The positional isomers of the G1 glycopeptide have been separated by UHPLC.


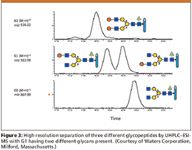
Figure 3
In biotechnology, the drug substance can be a complex mixture of complex glycoprotein structures, often not resolvable by any common analytical or MS techniques. To fully characterize each and every single, individual glycoprotein present is often not practical, and might just be impossible. It is clearly possible, for individual glycopeptides, first resolved by UHPLC and HPLC, to derive the entire glycan sequence, often on trace amounts of material injected. An alternative approach for the partial characterization of a mixture of glycoproteins or glycopeptides is illustrated in Figure 4, where N- and O-glycans have first been released from the glycoproteins, separated from the residual proteins, and then characterized by a variety of possible, analytical methods.



Figure 4
Thus, the full and complete chemical characterization of many biotech products on the market today was never possible before, might not be possible today, and might never be possible in the future. Recognition of this fact in the late 1990s led to coining of the phrase "well-characterized biological product" mentioned earlier (14). These meetings are still ongoing, and they have proven very useful, bringing together the entire biotech industry, FDA, and USP to try and resolve lingering or new problems related to new drug approvals (NDA), abbreviated new drug approvals (ANDA), and investigational new drug approvals (IND) (14,18).
However, WCBPs often cannot be fully characterized or quantitated, and therefore, analytical methods are validated using somewhat different approaches than for low molecular weight, conventional pharmaceuticals. (19,20,33). They are characterized chemically as best as possible, given the current limitations in analytical methods, technologies, and instrumentation. The fact that complex mixtures of glycoproteins, for example, have perhaps never been characterized fully, begs the following question: How can a biosimilar or biogeneric (these might not be identical) drug product ever be shown chemically equivalent to the proprietary product, without first requiring partial or full clinical testing for safety and efficacy? This is a thorny question that remains to this day a vexing issue for our FDA to resolve (1,4,6,14,15,40).
Protein size, shape, and complexity: Quite naturally, most biotech-derived drug substances, especially peptides and proteins, are considerably larger than their small molecule, conventional pharmaceuticals (for example, aspirin, acetominophen, and ibuprofen). Their complexity is not just in the sequence of their amino acids, number of chains, disulfide bridges, and other gross, three-dimensional (3D) features, but especially as regards their PTMs (7,14,17,40). These could be amino acid oxidations, deamidations, glycations, glycosylations, amino acid substitution, disulfide scrambling, or several hundred other, possible modifications (for example, loss of C-terminal Lys, formation of pyroglutamate at the N-terminal Glu). And, as alluded to earlier, today we usually are dealing, in most biotech products, with several variants or isoforms, as these often are termed, but they are all part of the final, total drug substance (14,17,40). And, if they are efficacious and safe, then they cannot or need not be removed from the final drug product. All of these variants are the total drug substance and eventually, after formulation, the final drug product. So, in devising and applying analytical procedures for newer biotech products, we need to consider that these often are very large, high molecular weight (MW) biopolymers of varying degrees of complexity. This often makes their analyses challenging and difficult, time-consuming, expensive, and incomplete for complete chemical characterizations. It is truly much easier to introduce to market a small molecule, low MW pharmaceutical than a large biopolymer of often very high MW (for example, 150 kDa).



Figure 5
Impurities in biopharmaceutical products: Impurities often can occur in conventional, low MW pharmaceuticals, but when they arise in biotech products, they are usually more difficult to remove (21). Impurities are usually of two types: drug-substance related and process related, arising from how the original drug substance was purified after expression and removal from the expression systems (ICH guideline). Or, in the process of purification, via chromatography, filtration, sedimentation, and other steps, impurities can be introduced (for example, protein A). Figure 5 illustrates the possible product- and process-related impurities that might be present in a commercial biopharmaceutical manufactured today. Impurities technically are not related to the drug substance in its primary amino acid sequence and often come from the cell expression system, such as host cell DNA, host cell proteins (HCP), host cell biological macromolecular complexes, and others. These must be fully removed (<0.1% relative peak areas) from the final drug substance before its chemical characterization and shown absent by more than one, single separation–detection scheme, whatever that might require. Then, there are the drug process-related impurities, not arising from the host cell, but rather somewhere along the process purification steps. These could be protein A or G from an affinity step, ligands from a reversed-phase or ion-exchange step, salts from a precipitation or solubilization step, and others. These too must be shown absent from the final drug substance after large-scale purification and before formulation. The drug product, as the final formulation, also must be shown devoid of impurities coming from the formulation materials. Basically, the only things in the final formulation should be the drug substance or substances and the impurity-free, formulation additives. This is often a challenging task, as evidenced by the recent problems that Genzyme has faced, with two of its already commercialized products, Cerazyme and Fabrazyme (22). In one earlier instance, a virus had entered the process-scale manufacturing site (then into the drug products), and in the other, small particles and fibers were found in drug products going to or gone to market. These are not desirable occurrences and need to be avoided, as much as humanly possible. FDA product recalls are often the final outcome of such serious instances of impurities being present in the final drug products. Or, FDA can hasten (fast track) the introduction of a competitive product, so as to provide the needed drug for patients who had been using a drug just denied access to or taken from the market (23). Table II describes how, in part, impurities commonly found in protein pharmaceuticals might be analyzed, utilizing routinely available analytical techniques.


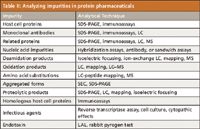
Table II: Analyzing impurities in protein pharmaceuticals
3D Structures
Though low MW, conventional pharmaceuticals also have 3D structures, they are not nearly as complex and complicated as for large MW antibodies or glycoproteins. However, with biopolymers, there are often some, perhaps not all, 3D structures that exhibit the desired biological efficacy in animals and humans, and are entirely safe when properly administered. Most biologics, another term for biopharmaceuticals, are administered parenterally, by injection, rather than taken orally or by inhalation. They just do not usually survive the rigors of the human intestinal system when taken orally, and so prove useless for any successful treatments. Biopolymers have several possible 3D structures (actually proteins have four possible levels of structure, but the differences between levels 3–4 are not pertinent to the discussion here) (11–13,24,25). Only some of these possible 3D structures have the desired biological properties, and the others are basically useless or even dangerous to the patient (for example, denatured, immunogenic aggregates). During expression, cleanup, or process-scale purification steps, 3D structures can and often do change from active to inactive or denatured or aggregates. The ultimate proof of the desired 3D structure is retained biological activity, safety, and chemical identification by FTIR, high resolution NMR spectroscopy, X-ray crystallography, low-angle laser light scattering spectroscopy (LALLS) or MALS, viscometry, analytical ultracentrifugation, optical rotatory dispersion (ORD), circular dichroism (CD), H/D exchange MS, IMS-MS, and other, even more advanced analytical techniques (8–11,26). Clearly, it is not enough, when doing complete chemical characterization of a new biotech antibody or other glycoproteins, just to show it has the expected sequence of amino acids from the DNA expression system, or glycans attached at the right N/O sites. In addition, one must also show the correct shape or 3-D structures. These are not always simple requirements.
Where Is the Biotechnology Industry Today and Where Is It Going Tomorrow?
This, too, is an area for future columns. Here, we would like to state the major classes of biotechnology products being marketed and researched today (1,3,4,6):
- Glycoproteins
- Fusion proteins
- Antibodies
- Synthetic, smaller peptides, and
- Complex mixtures of polypeptides.
These appear to comprise at least 95% of currently marketed biotech products, and especially today, the recombinant, human monoclonal antibodies (rhMAbs) appear to be garnering the bulk of INDs and NDAs around the world. Most biotech products are being expressed in animal, mammalian cell systems, but there are some very good examples of plant-expressed glycoproteins coming to market for human and animal applications (23). Surely, the future most likely will mimic this (partial) listing of where the biotech industry is today and where it is going in the near future with regard to the types of products being generated and successfully marketed (Table I).
The Future of "Biotechnology Today"
In finishing this first installment of a new column, we naturally think of topics for future installments. We wish to enlist your interests and needs, and though future column topics are plentiful — for example, protein expression systems, characterization of PTMs, biotech impurity profiling, process scale purifications, and countless others — we welcome your constructive input and suggestions. We very much want these columns to be relevant and of interest to those working or hoping to work in the biotech industry, as well as academic and regulatory scientists, students and advisors, and managers. Please e-mail us with your thoughts and ideas. We promise to give them all serious consideration and acknowledgment. If anyone out there would like to work with us or others on future such columns, we welcome those opportunities also. It would be great to have experts from within the biotech world contribute to future columns, with or without our involvement. And, with that invite, we end this first column of what, hopefully, will be a very long-lived column series in LCGC.
Ira S. Krull "Validation Viewpoint" Co-Editor Ira S. Krull is an Associate Professor of chemistry at Northeastern University, Boston, Massachusetts, and a member of LCGC's editorial advisory board.
Anurag S. Rathore is a biotech CMC consultant and an associate professor with the Department of Chemical Engineering, Indian Institute of Delhi, India. He is also a member of BioPharm International's editorial advisory board. Direct correspondence to: lcgcedit@advanstar.com
References
(1) The Future of Biotech, The 2010 Guide to Emerging Markets and Technology (BioWorld Today Publishers, 2010).
(2) Massachusetts Biotechnology Council, Cambridge, MA, http://massbio.org
(3) BIO, Biotechnology Industry Organization, www.BIO.com
(4) Time, November 2, 2009, pp. 36–39.
(5) BioTerminology, a Guide to the Biopharmaceutical Lexicon, First Edition, (Waters Corporation, Milford, Massachusetts, February, 2009).
(6) Scientific American WorldView, A Global Biotechnology Perspective, The Future of Biotech, The 2010 Guide to Emerging Markets and Technology (BioWorld Today, Scientific American, Inc., Publishers, 2009).
(7) C.T. Walsh, Posttranslational Modification of Proteins. Expanding Nature's Inventory (Roberts and Company, Publishers, Englewood, Colorado, 2006).
(8) State-of-the-Art Analytical Methods for the Characterization of Biological Products and Assessment of Comparability, A.R. Mire-Sluis, Ed., Developments in Biologicals, volume 122 (Karger, Basel, CH, 2005).
(9) G. Marko-Vargy and P. Oroszlan, Emerging Technologies in Protein and Genomic Material Analysis (Elsevier Science Publishers, Amsterdam, The Netherlands, 2003).
(10) Analytical Techniques for Biopharmaceutical Development, R. Rodriguez-Diaz, T. Wehr, and S. Tuck, Eds. (Marcel Dekker, Taylor & Francis Group, New York, 2005).
(11) R. Kellner, F. Lottspeich, and H.E. Meyer, Microcharacterization of Proteins, Second Edition (Wiley-VCH, Weinheim, Germany, 1999).
(12) U. Langel et al., Introduction to Peptides and Proteins (Taylor & Francis Group, CRC Press, Boca Raton, Florida, 2010).
(13) R.L. Lundblad, The Evolution from Protein Chemistry to Proteomics, Basic Science to Clinical Applications (Taylor & Francis Group, CRC Press, Boca Raton, Florida, 2006).
(14) WCBP Meetings, California Separations Society (CaSSS), website: www.casss.org (January 25-27, 2010, Washington, DC)
(15) US Food & Drug Administration, Rockville, Maryland. www.fda.gov/
(16) European Medicines Agency, London, UK. www.emea.europa.eu/
(17) Z. Shriver, S. Raguram, and R. Sasisekharan. Nature Reviews, Drug Discovery 3, 863 (2004).
(18) California Separations Society, Emeryville, California. www.casss.org/
(19) I.S. Krull, P.T. Kissinger, and M.E. Swartz, LCGC 26(11), 1110–1117 (2008).
(20) M.E. Swartz and I.S. Krull, LCGC 27(7), 550–557 (2009).
(21). M. Swartz and I. Krull, LCGC 25(12), 1196–1201 (2007).
(22) The Boston Globe, Saturday, November 14, 2009 edition, www.bostonglobe.com.
(23) Protalix, Ltd., Carmiel, Israel, www.protalix.com
(24) D. Whitford, Proteins — Structure and Function (J. Wiley & Sons, Ltd., Chichester, UK, 2005).
(25) T.E. Creighton, Proteins- Structures and Molecular Properties, Second Edition (W.H. Freeman & Co., New York, 1993).
(26) Handbook of Instrumental Techniques for Analytical Chemistry, F.A. Settle, Ed. (Prentice Hall PTR, Upper Saddle River, New Jersey, 1997).
(27) P. Olivova, W. Chen, A.B. Chakraborty, and J.C. Gebler, Rapid Commun. Mass Spectrom. 22, 29–40 (2008).
(28) B.S. Kendrick, G. Chrimes, S.L. Cockrill, J.P. Gabrielson, K.K. Arthur, B.D. Prater, Q. Qin, B. Zhang, and A.S. Rathore, BioPharm Intl., 32–44 (August, 2009).
(29) A.S. Rathore and H. Winkle, Nature Biotech. 26, 26–34 (2009).
(30) A.S. Rathore, Trends in Biotech. 27, 546–553 (2009).
(31). M.A. Schnerman, B.R. Sunday, S. Kozlowski, K. Webber, H. Gazzano-Santoro, and A. Mire-Sluis, Bioprocess Int., 42–52 (February 2004).
(32) I.S. Krull and M.E. Swartz, "Validation in biotechnology and well-characterized biopharmaceutical product," in Pharmaceutical Regulatory Guidance Book, pp. 18–23 (Advanstar Communications, Woodland Hills, California, 2006).
(33) I. Apostol and D. Kelner, BioProcess International, Part 1, 12–19 (September, 2008) and Part 2, 12–19 (October, 2008).
(34) X. Chen and G.C. Flynn, Anal. Biochem. 370, 147–161 (2007).
(35) M. Kinter and N.E. Sherman, Protein Sequencing and Identification Using Tandem Mass Spectrometry (Wiley-Interscience Publishers, John Wiley & Sons, Inc., New York, 2000).
(36) C. Dass, Principles and Practice of Biological Mass Spectrometry (Wiley-Interscience Publishers, John Wiley & Sons, Inc., New York, 2001).
(37) L.C. Santora, K. Stainly, I.S. Krull, and K. Grant, Biomed. Chromatogr. 20, 843–856 (2006).
(38) GTC Biotherapeutics Corporation, Framingham, Massachusetts. http://www.genzymetransgenics.com/science.html
(39) G.W. Somsen and W.M.A. Niessen, J. Chromatogr., A 1159, 1–258 (2007).
(40) A.S. Rathore, Trends in Biotech. 27, 698–705 (2009).












AOAC International Awarded NIST Grant for Developing Drug Testing Standards
October 31st 2024The grant will be part of a new collaborative scientific initiative to address the need for standards that define the desired performance of lateral flow immunoassay test strips to detect illicit drugs in tablets and powders.


Supercritical Fluid Chromatography for Separating Chemotherapy Anti-Nausea Medication
October 30th 2024Scientists from Peking Union Medical College & Chinese Academy of Medical Sciences created a new method for separating palonosetron hydrochloride, which is frequently used to prevent acute or delated nausea and vomiting caused by chemotherapy.

HILIC Peptide Retention Times Predicted Using New Approach
October 29th 2024Manitoba Centre for Proteomics and Systems Biology scientists produced a new means of predicting peptide retention times for hydrophilic interaction liquid chromatography (HILIC) at acidic pH in formic-acid based eluents.



